




Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
Slides: https://slides.com/jbloom/grc2025
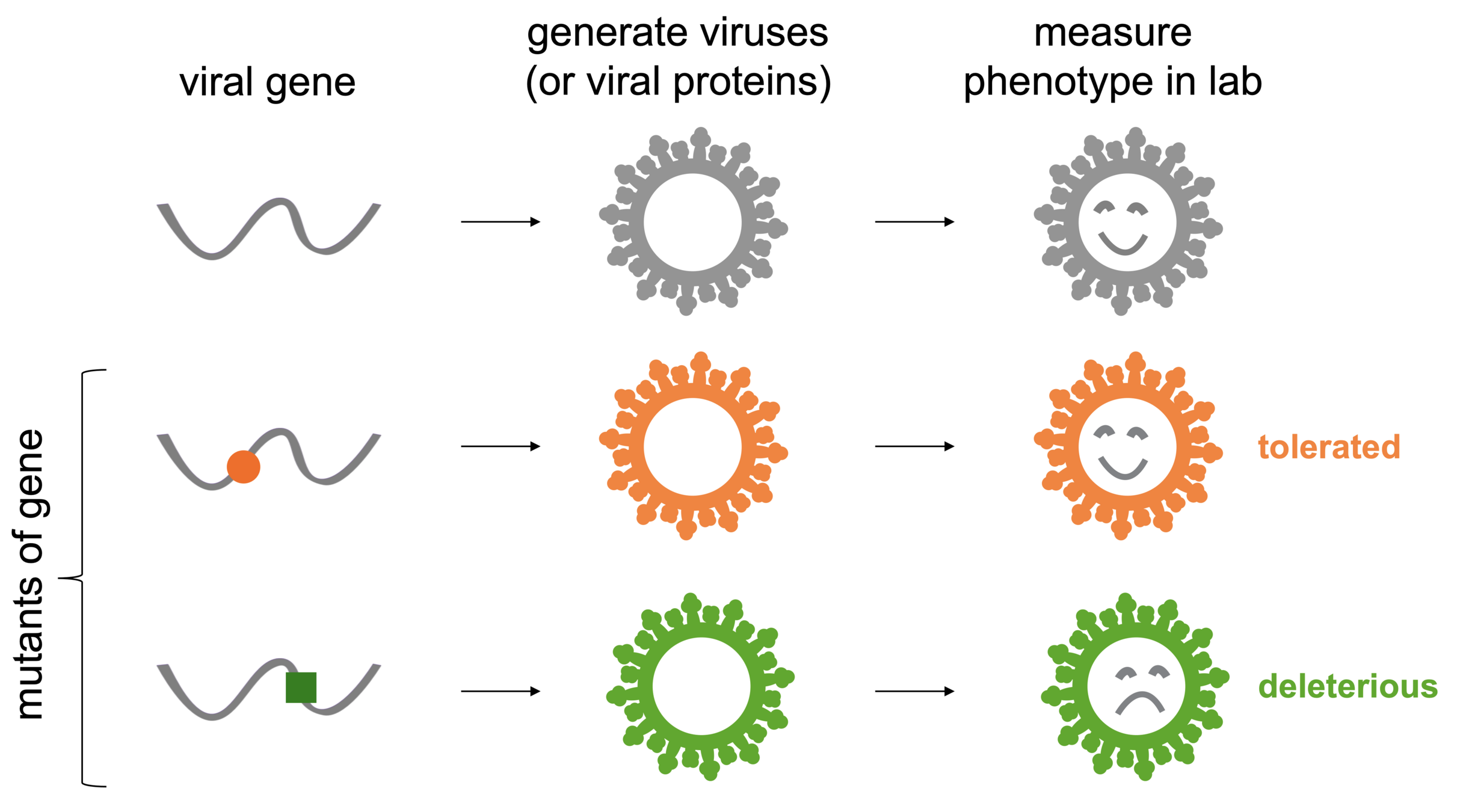
Yeast display or lentiviral pseudotype libraries allow us to measure many mutants at once by pooling them all together and reading out effects of mutations by deep sequencing (Starr et al, 2020; Dadonaite et al, 2023)
Laborious: even with deep mutational scanning, it's a lot of effort.
Laborious: even with deep mutational scanning, it's a lot of effort.
Lab assays measure effects of mutations in cells or mice, not humans. This is not the same as fitness in the real world.
Laborious: even with deep mutational scanning, it's a lot of effort.
Lab assays measure effects of mutations in cells or mice, not humans. This is not the same as fitness in the real world.
Some viral proteins have poorly understood functions that lack good lab assays.
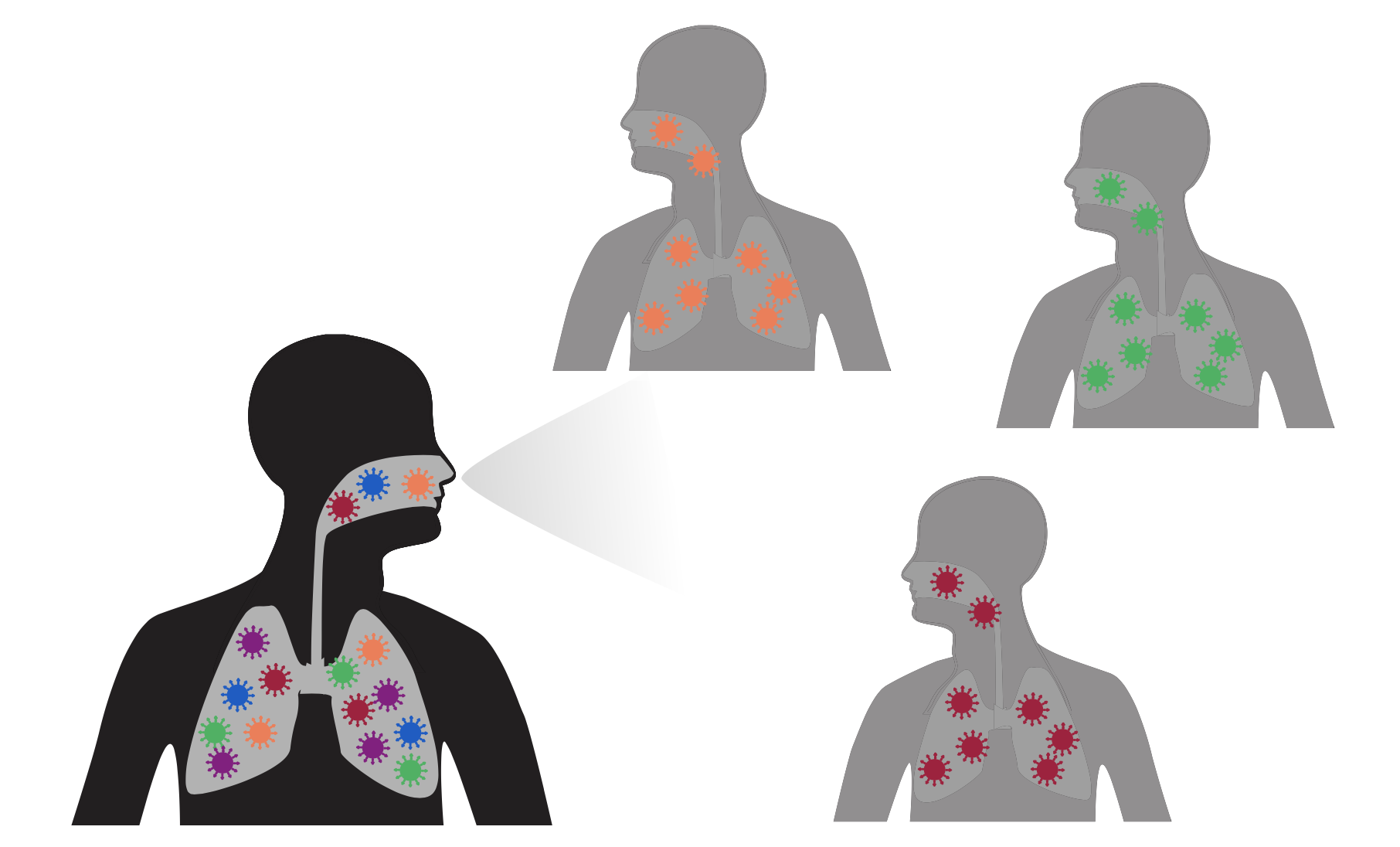
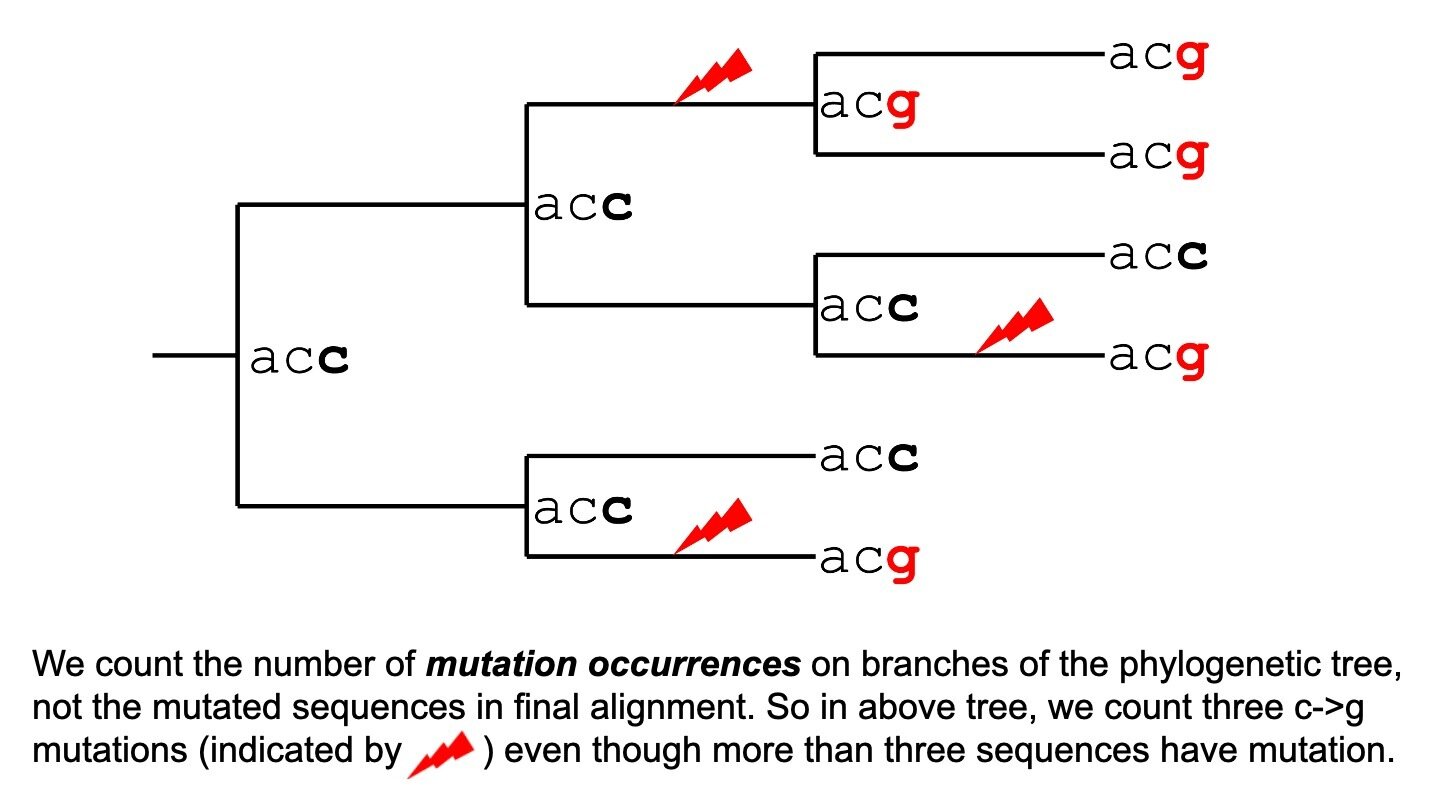
We calculate effect as log of actual versus expected mutation counts
fitness effect of mutation = log (actual counts / expected counts)
Effects of zero indicate neutral mutation, negative indicates deleterious mutation
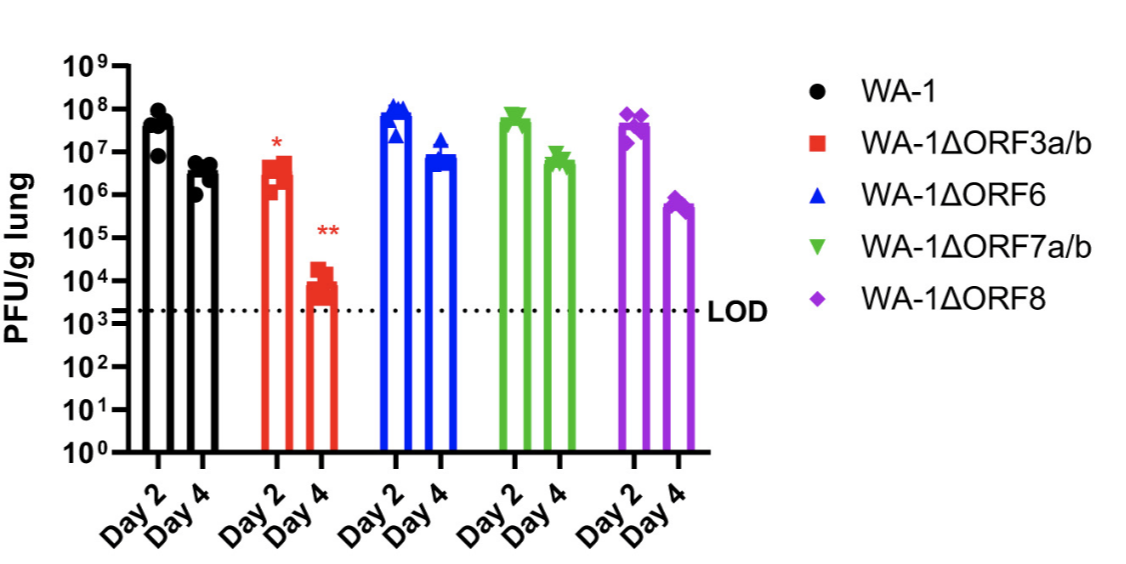
Experiments show that only accessory gene deletion that strongly attenuates virus in animal models is ORF3 (McGrath et al, 2022)
Two spike deep mutational scans using different underlying methodologies: lentiviral pseudotyping of spike or yeast display of RBD
Quantitative relationship between the ratio of observed versus expected counts and fitness depends on sampling intensity
Quantitative relationship between the ratio of observed versus expected counts and fitness depends on sampling intensity
There is additional information in dynamics of mutation after it occurs that our method currently does not leverage
Quantitative relationship between the ratio of observed versus expected counts and fitness depends on sampling intensity
There is additional information in dynamics of mutation after it occurs that our method currently does not leverage
Accuracy of our our approach depends critically:
Quantitative relationship between the ratio of observed versus expected counts and fitness depends on sampling intensity
There is additional information in dynamics of mutation after it occurs that our method currently does not leverage
Accuracy of our our approach depends critically:
This overall approach could be applied to many viruses / organisms with enough sequencing
Estimates of mutation rate
Kelley Harris, Annabel Beichman
Assistance with UShER
Angie Hinrichs, Russ Corbett-Detig
These slides: https://slides.com/jbloom/grc2025
By Jesse Bloom
Estimating effects of mutations to all SARS-CoV-2 proteins from actual versus expected mutation counts in natural sequences



