
























Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
These slides at https://slides.com/jbloom/vaxco-2025
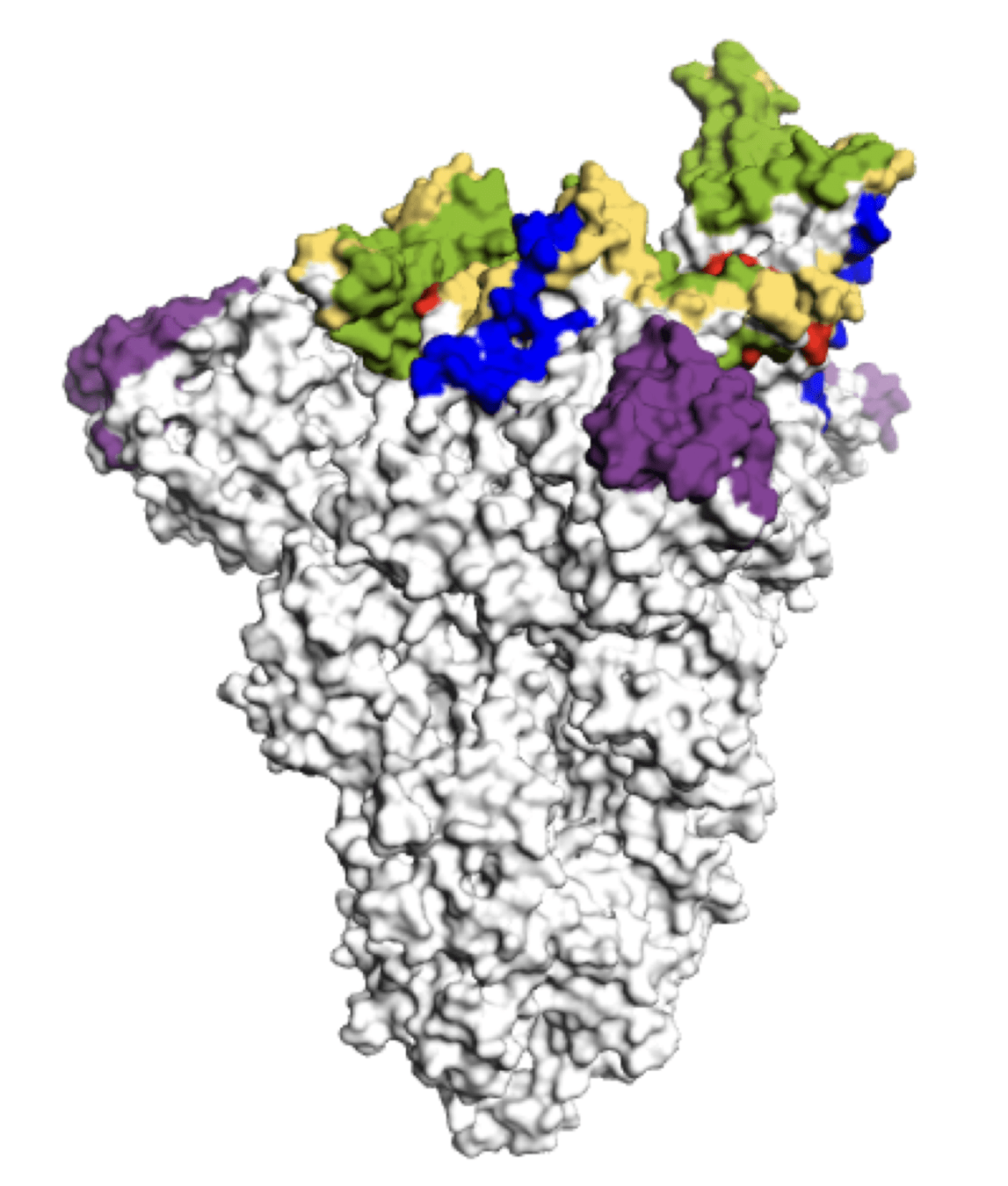
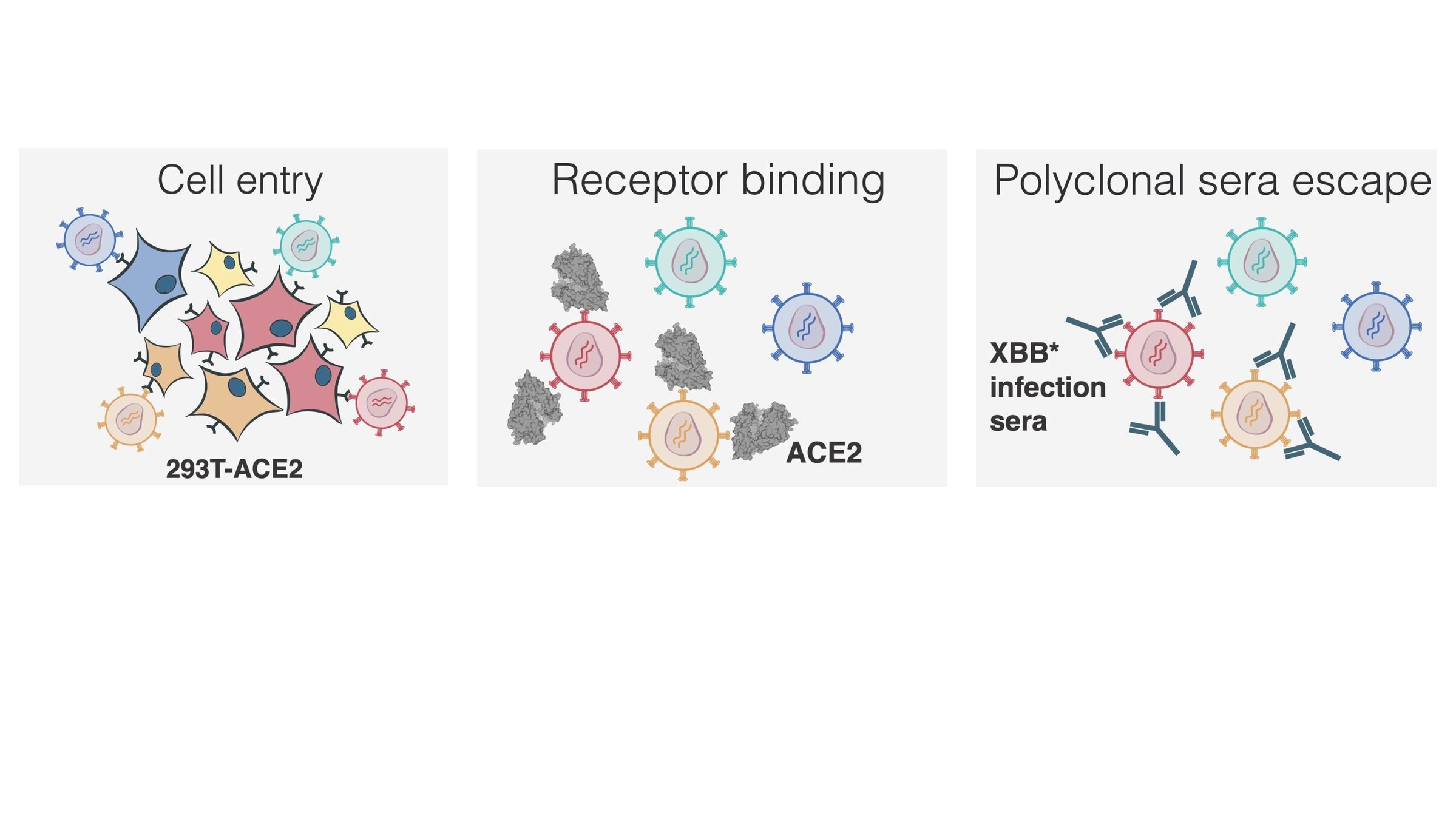
Main regions where neutralizing antibodies bind
Main regions where neutralizing antibodies bind
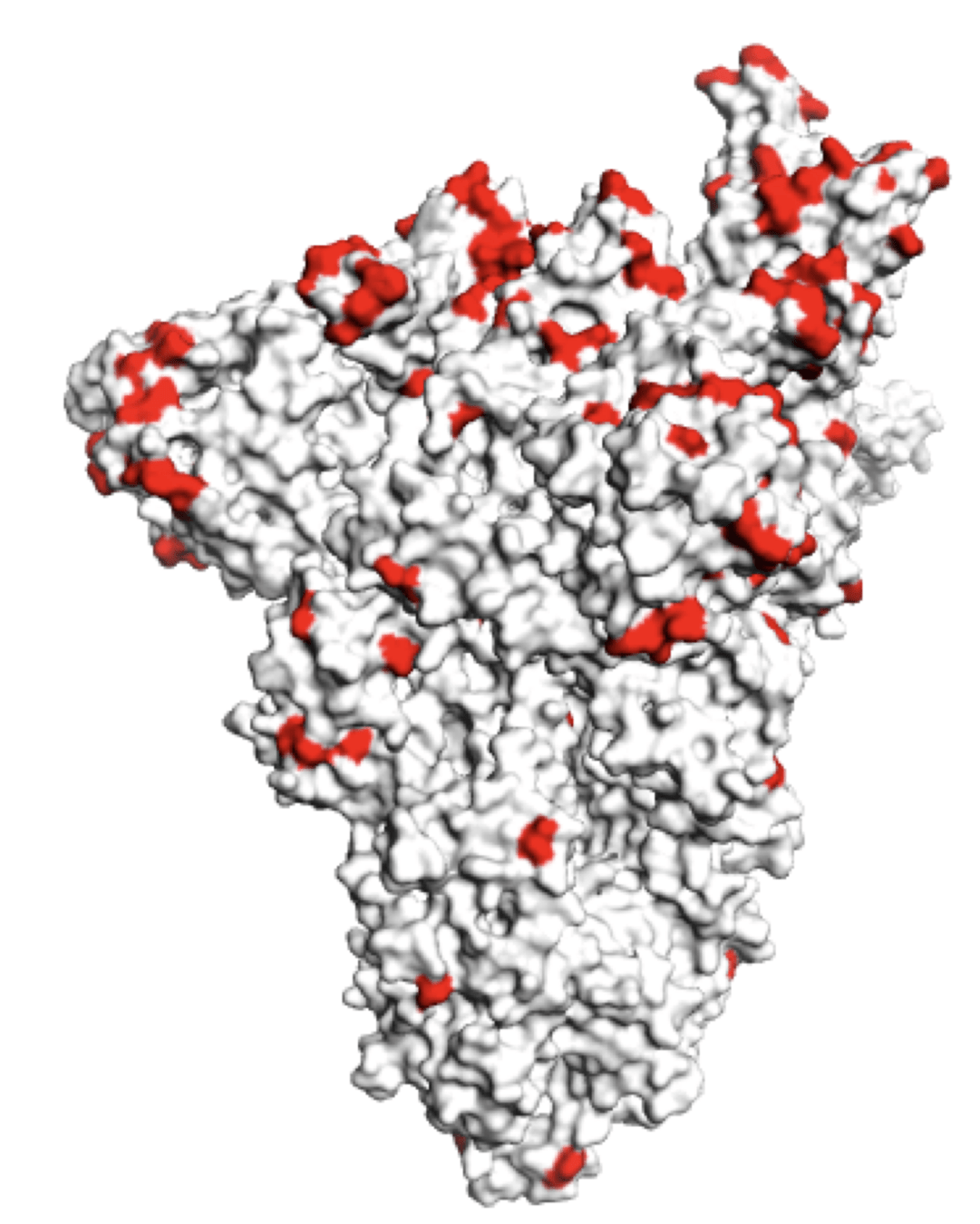
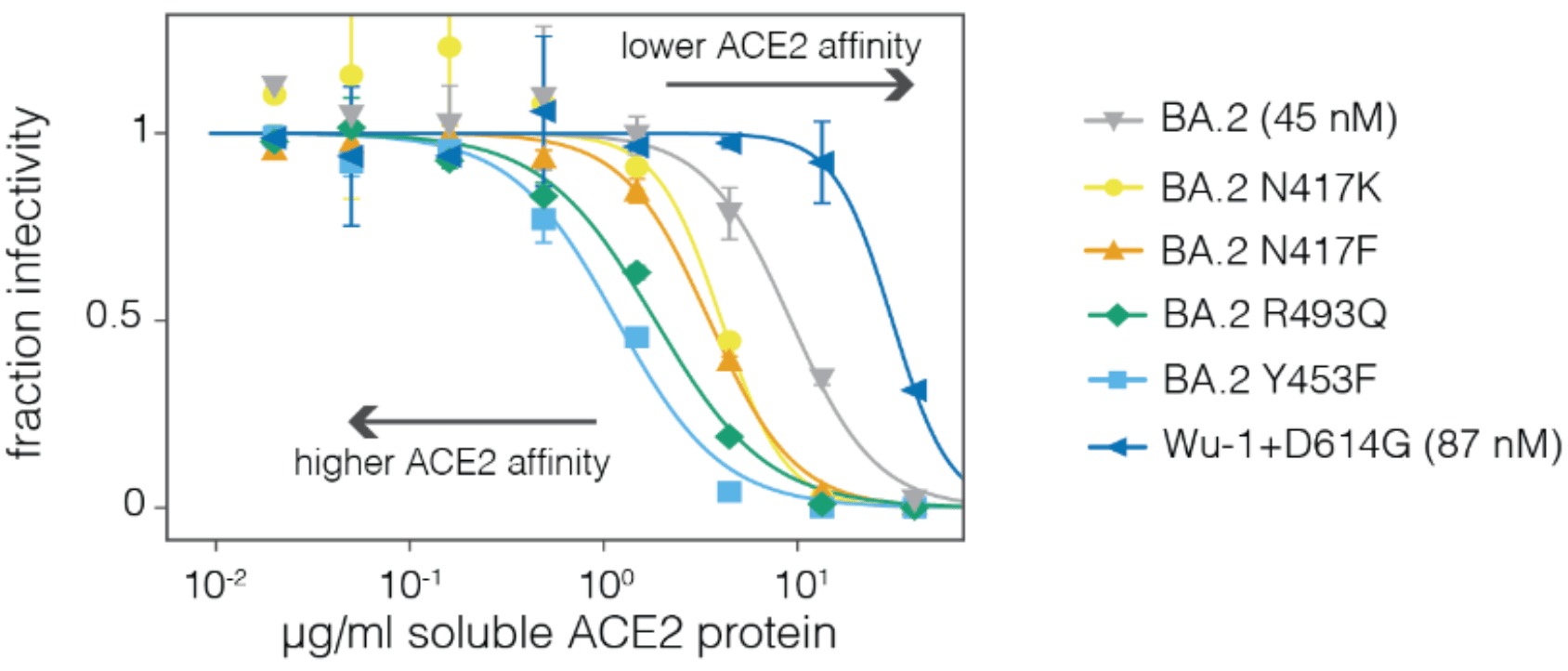
Sites of mutations in recent (BA.2.86) SARS-CoV-2 strain relative to early 2020 strain
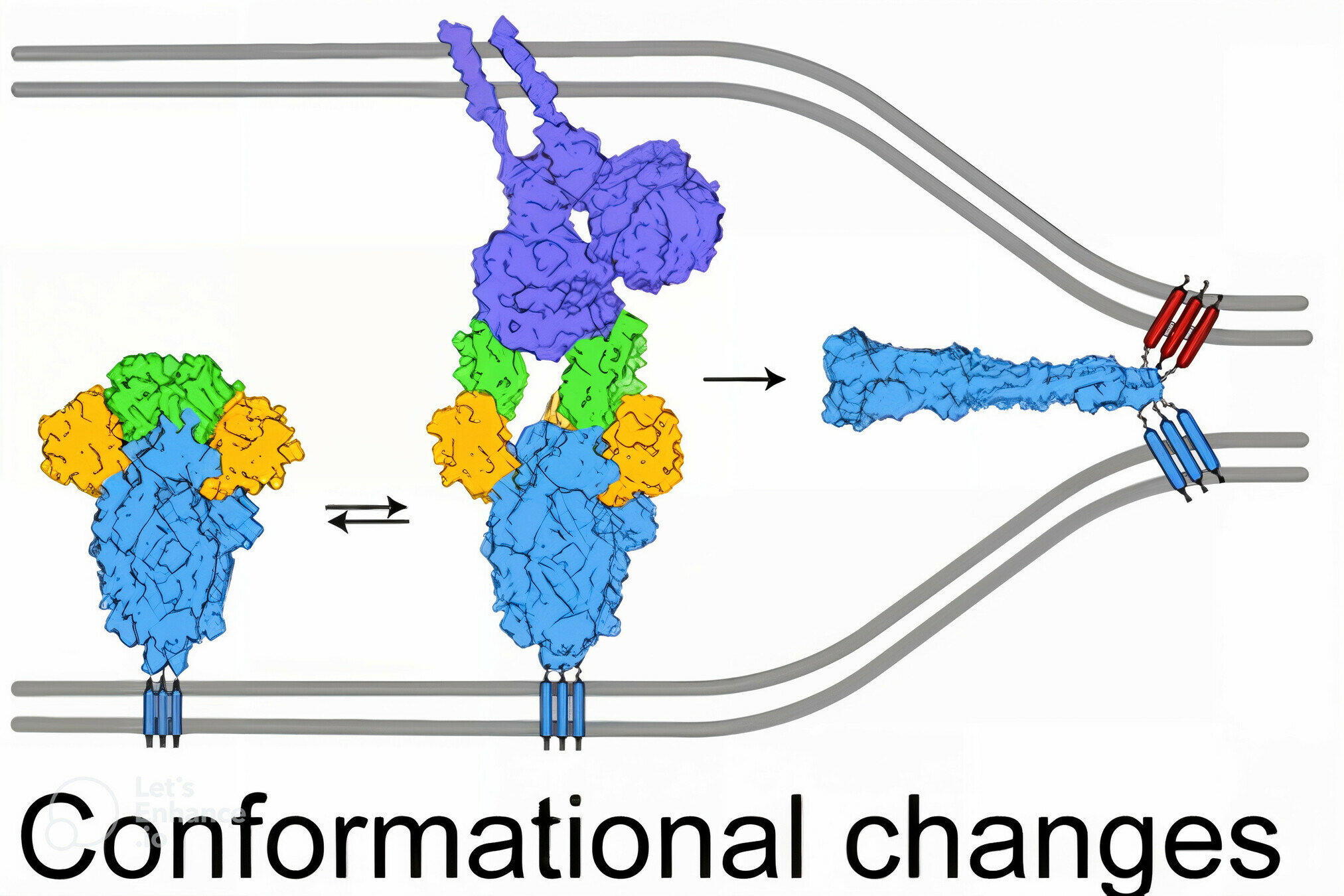
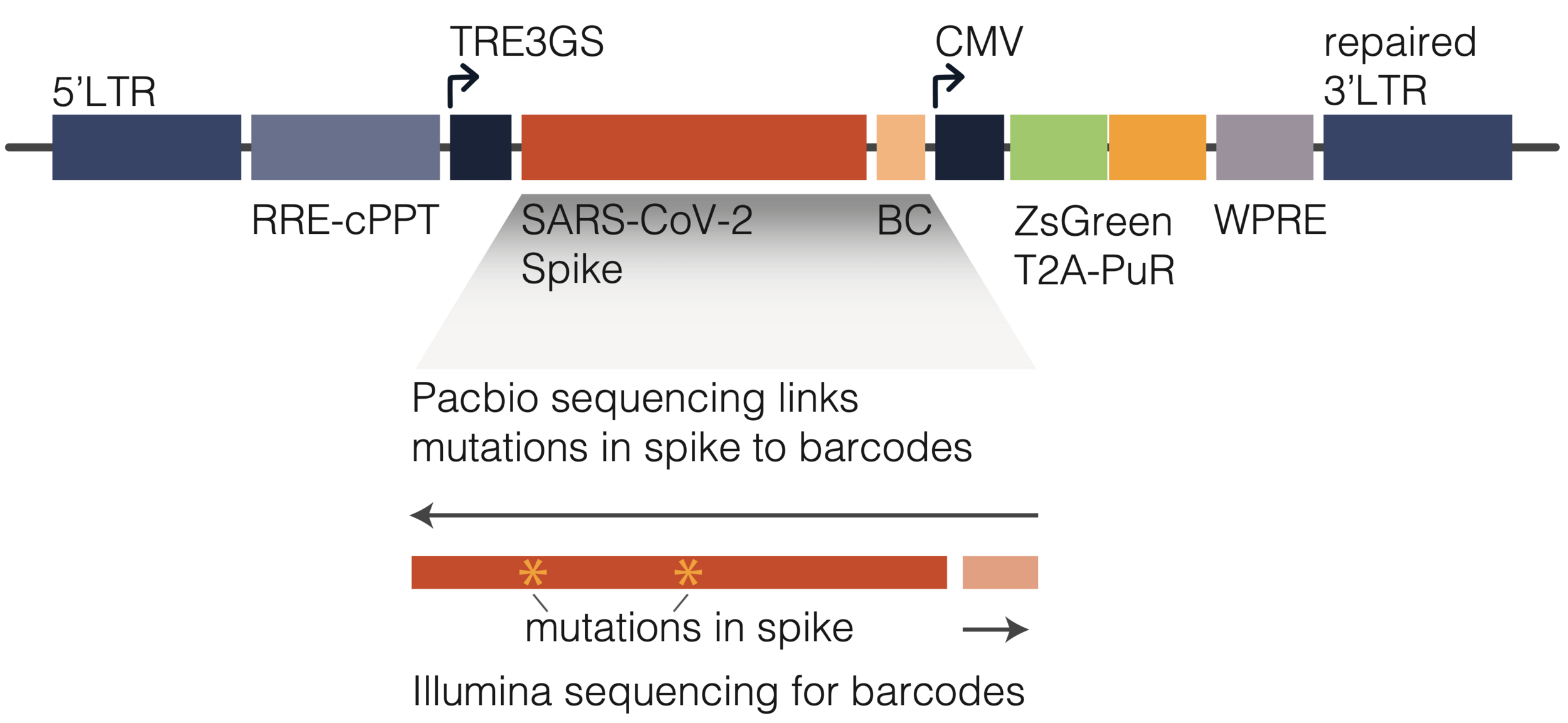
viral membrane
cell membrane
spike
spike conformational change
Image adapted from here
ACE2
antibody
Image adapted from here
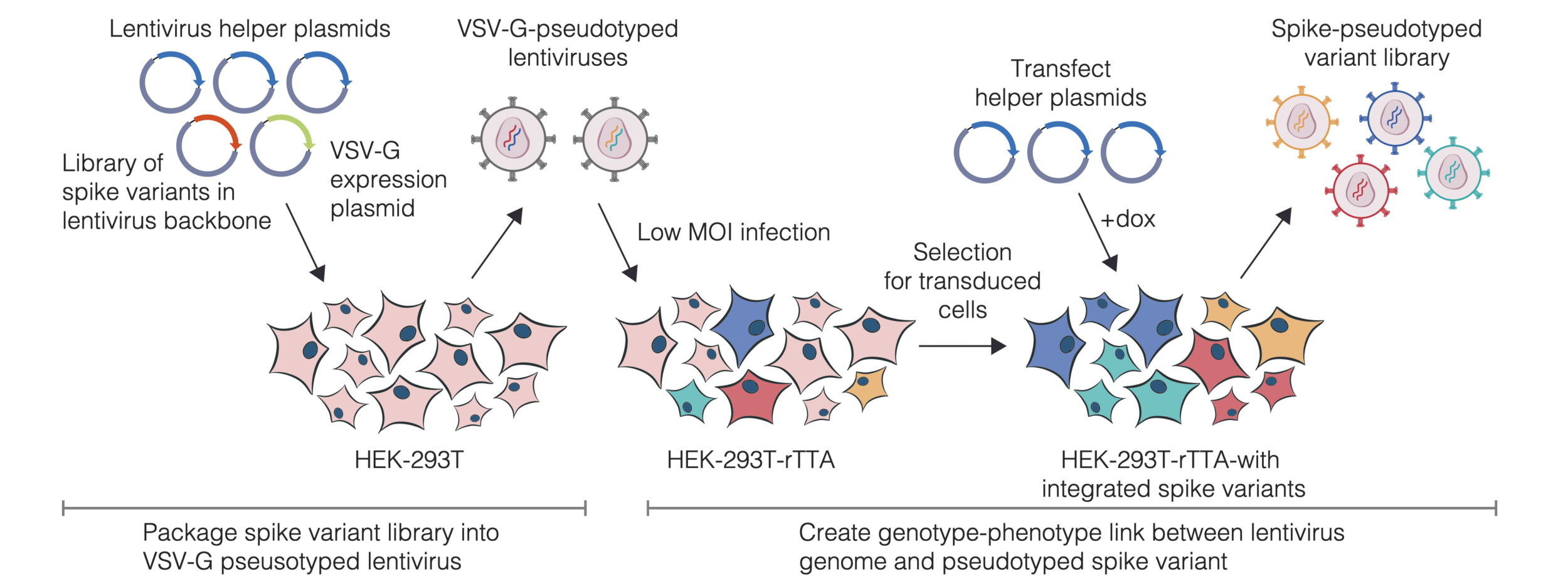
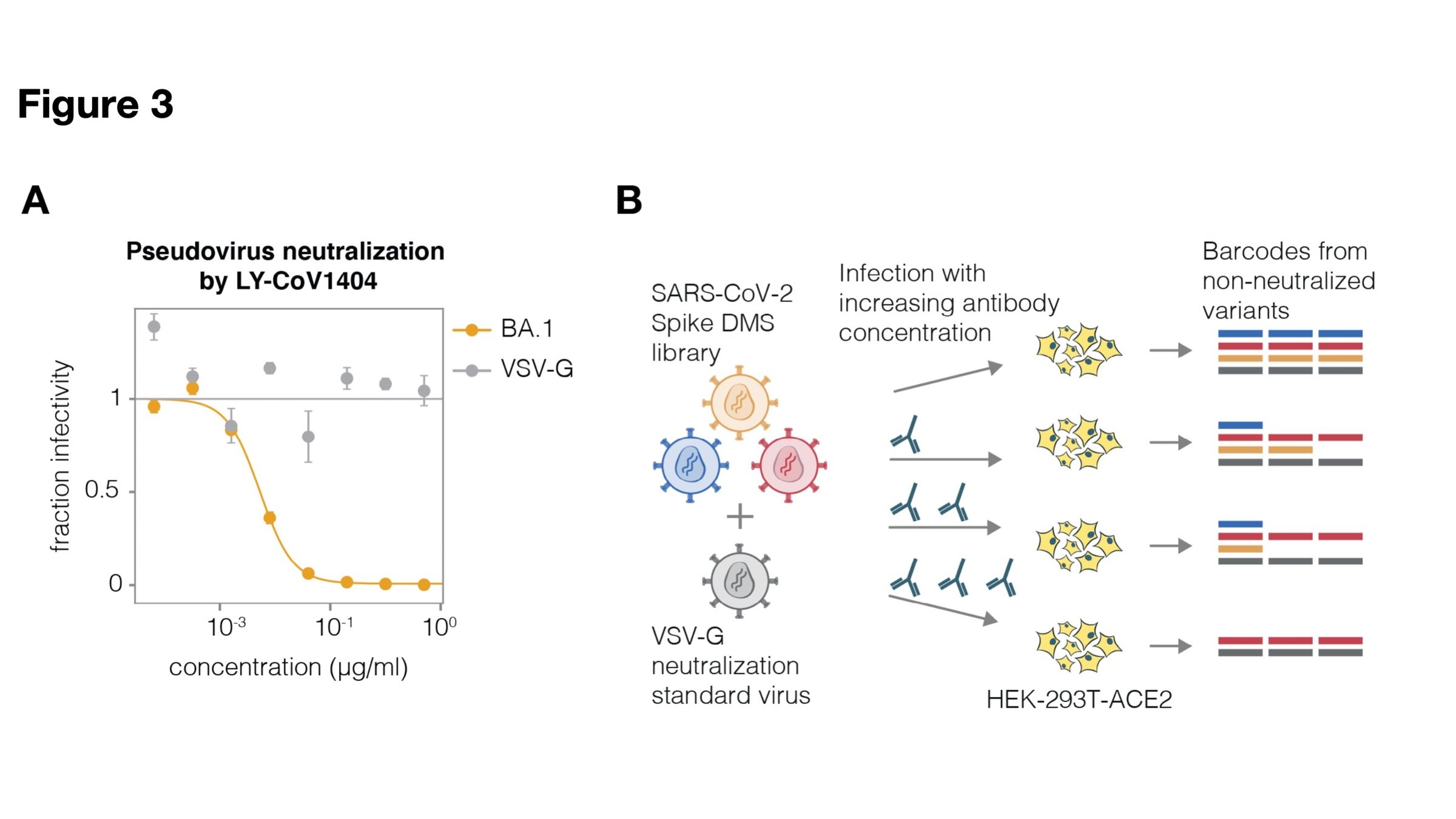
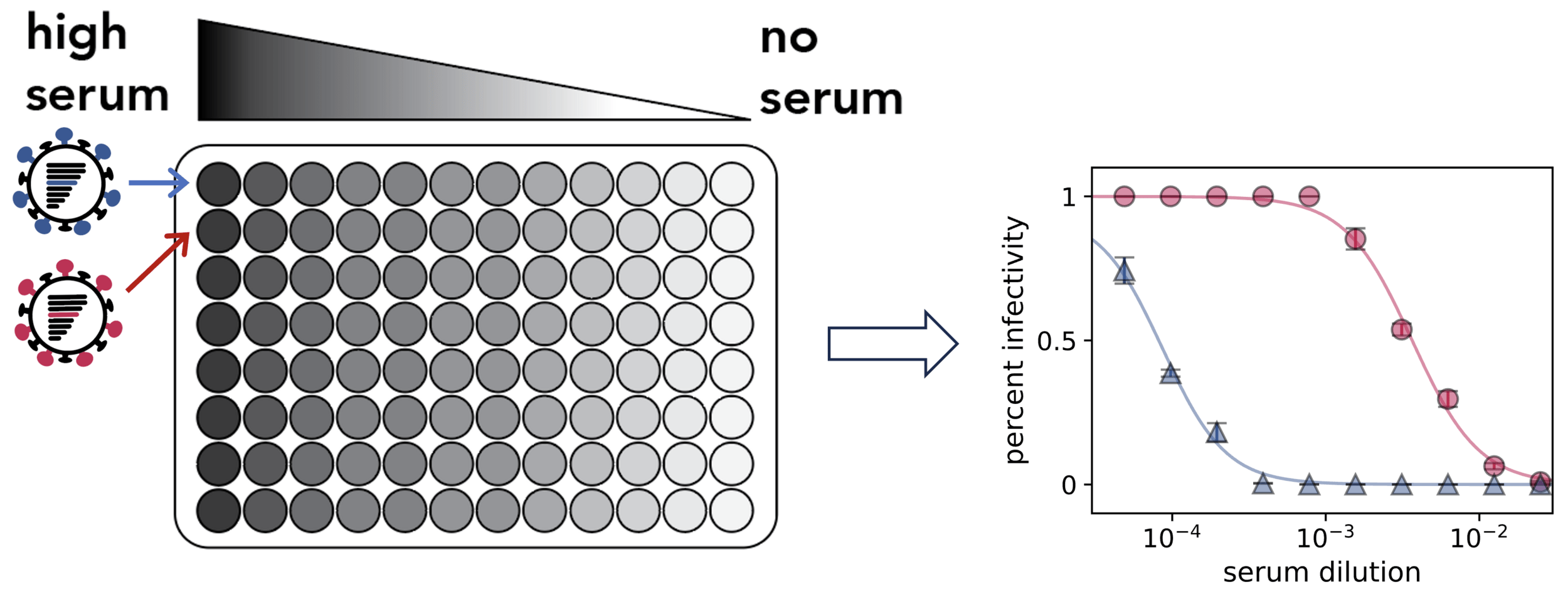
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
With Trevor Bedford & Ben Murrell
With Trevor Bedford & Ben Murrell
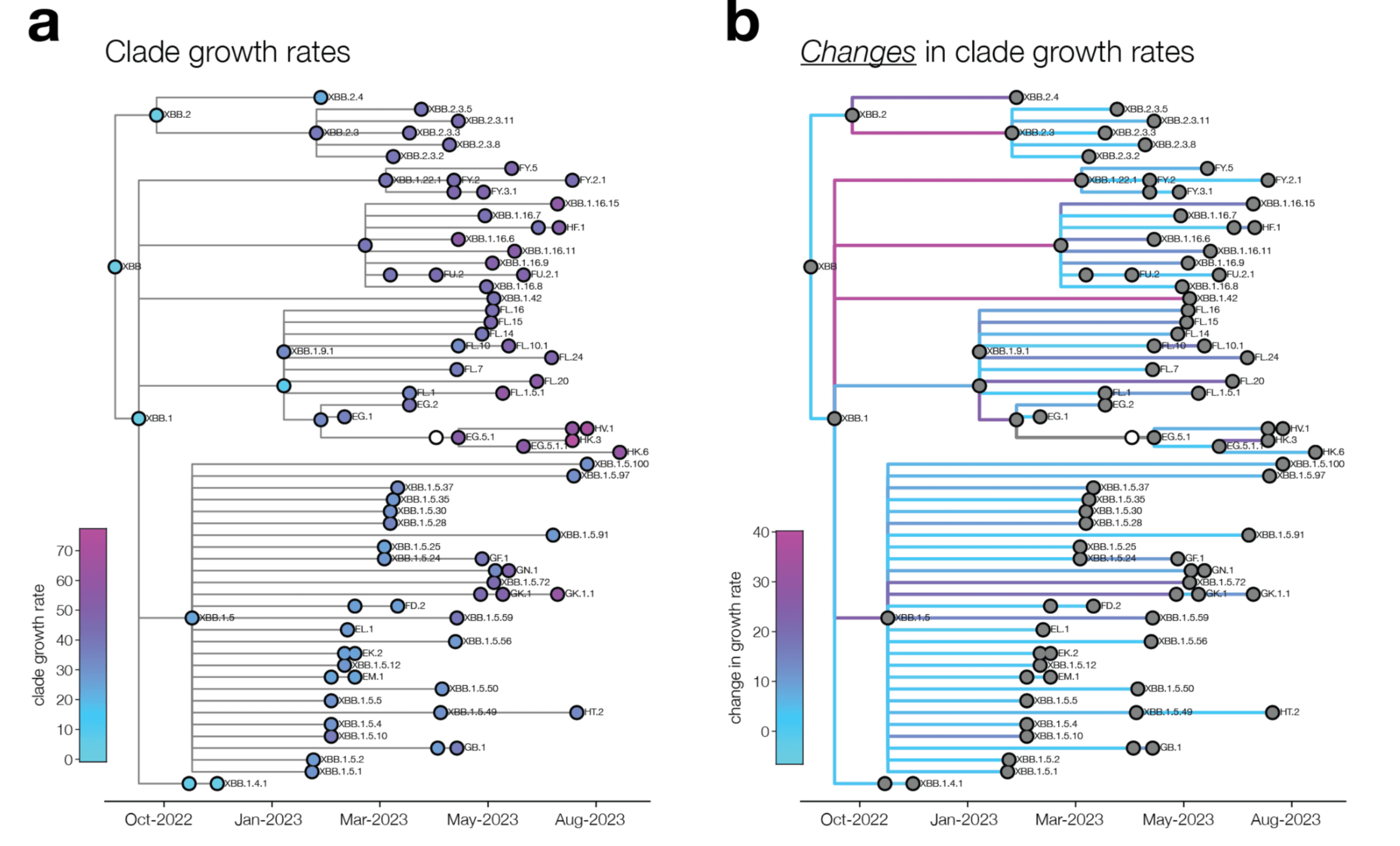
change in clade growth
clade growth
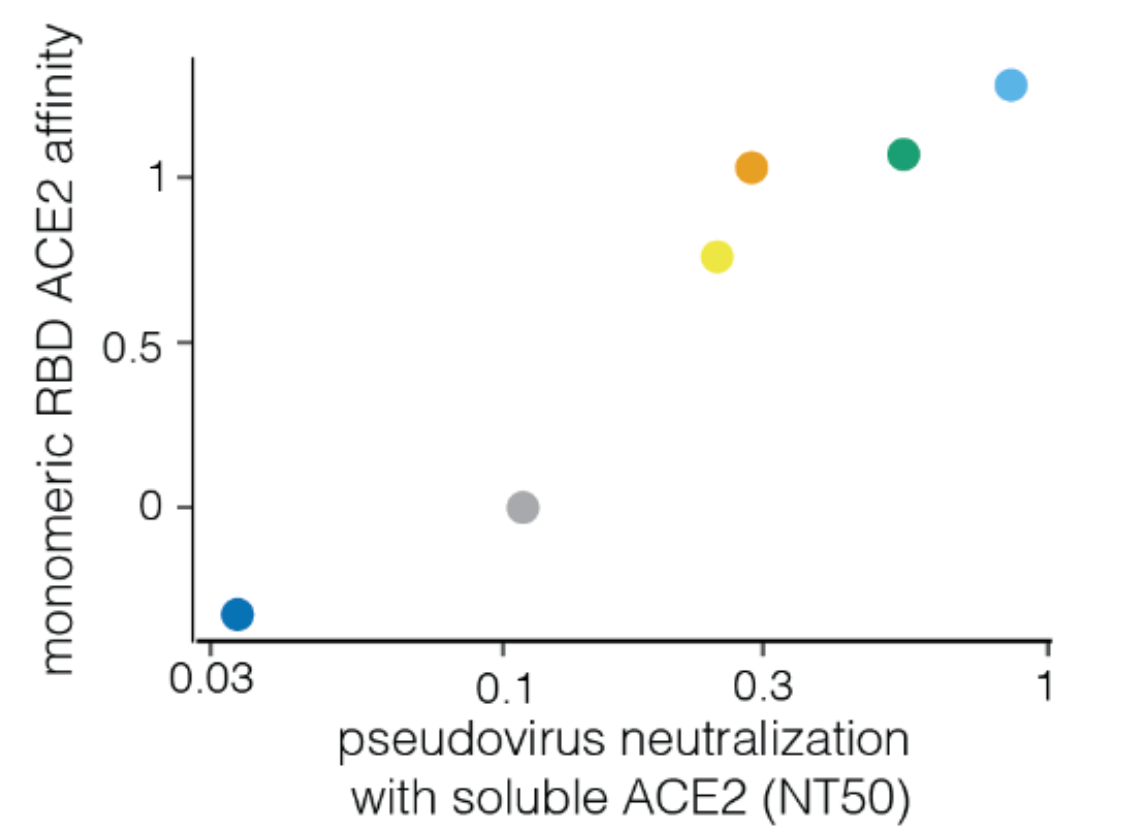
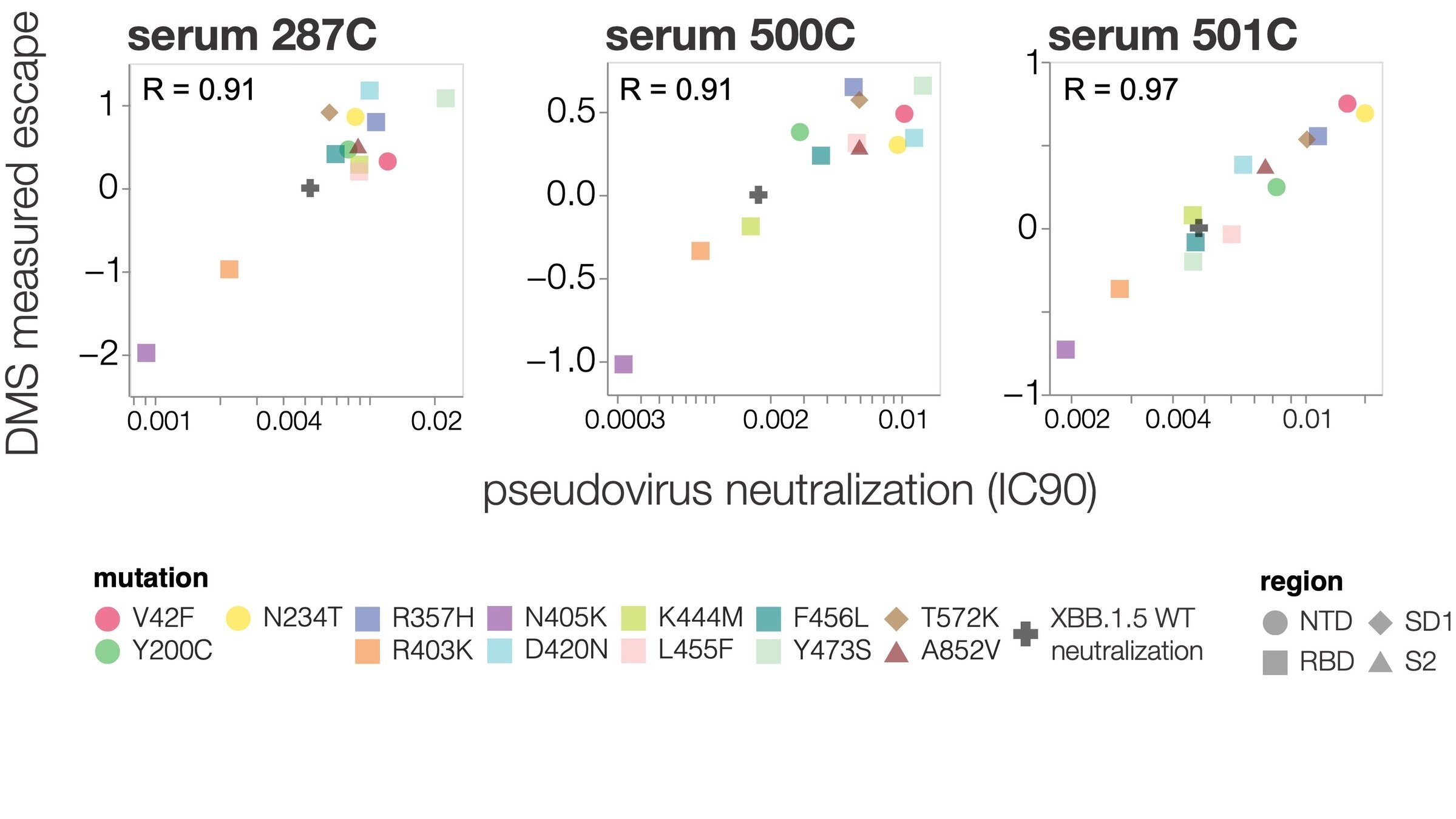
We can explain ~55% of the variance in growth of different clades, with largest fraction of variance uniquely explained by sera escape.
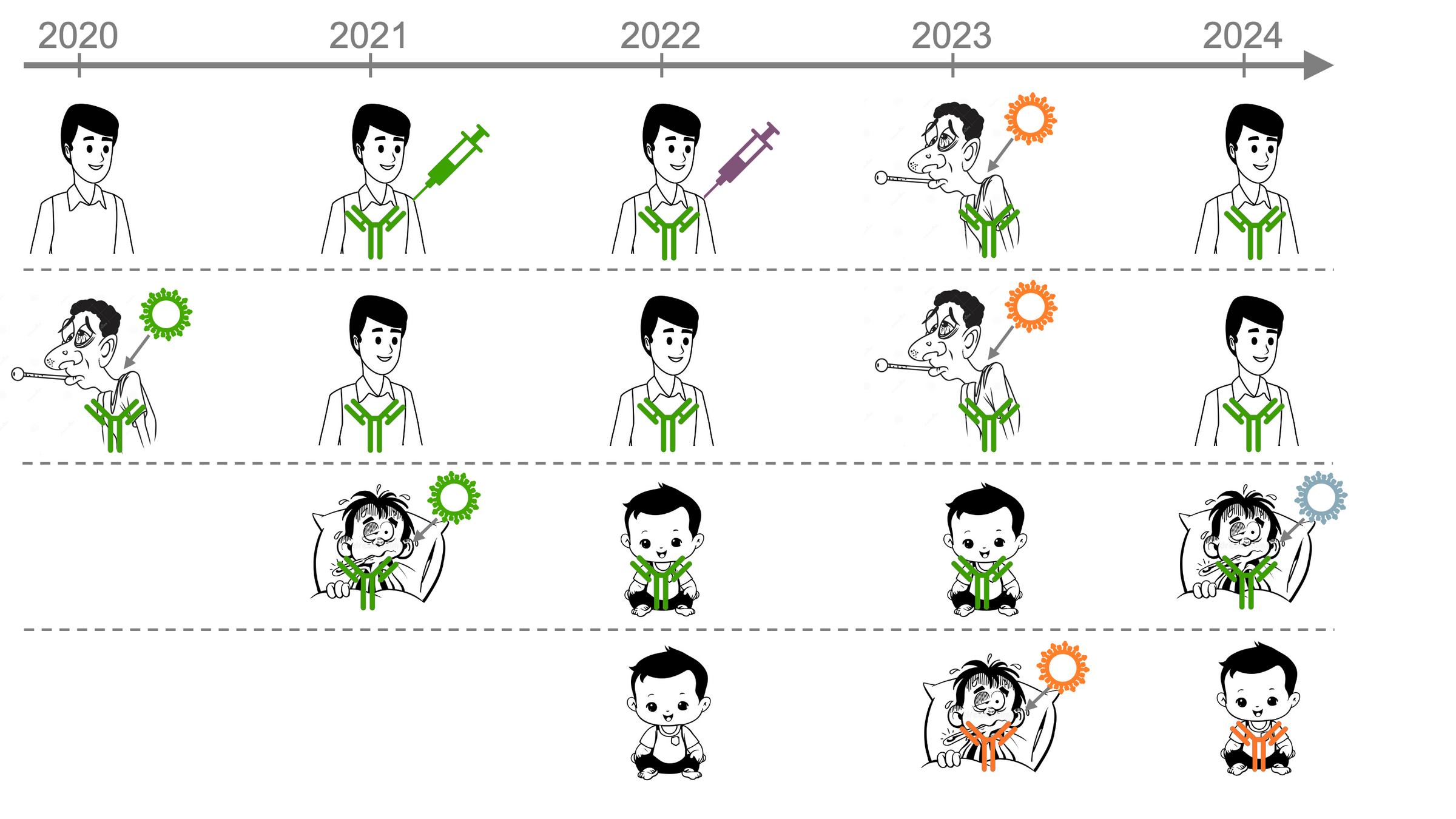
First exposed to early strain in original vaccine
First infected by an early strain (pre-Omicron)
Imprinted by recent strain
Adults vaccinated with original strain followed by various exposures
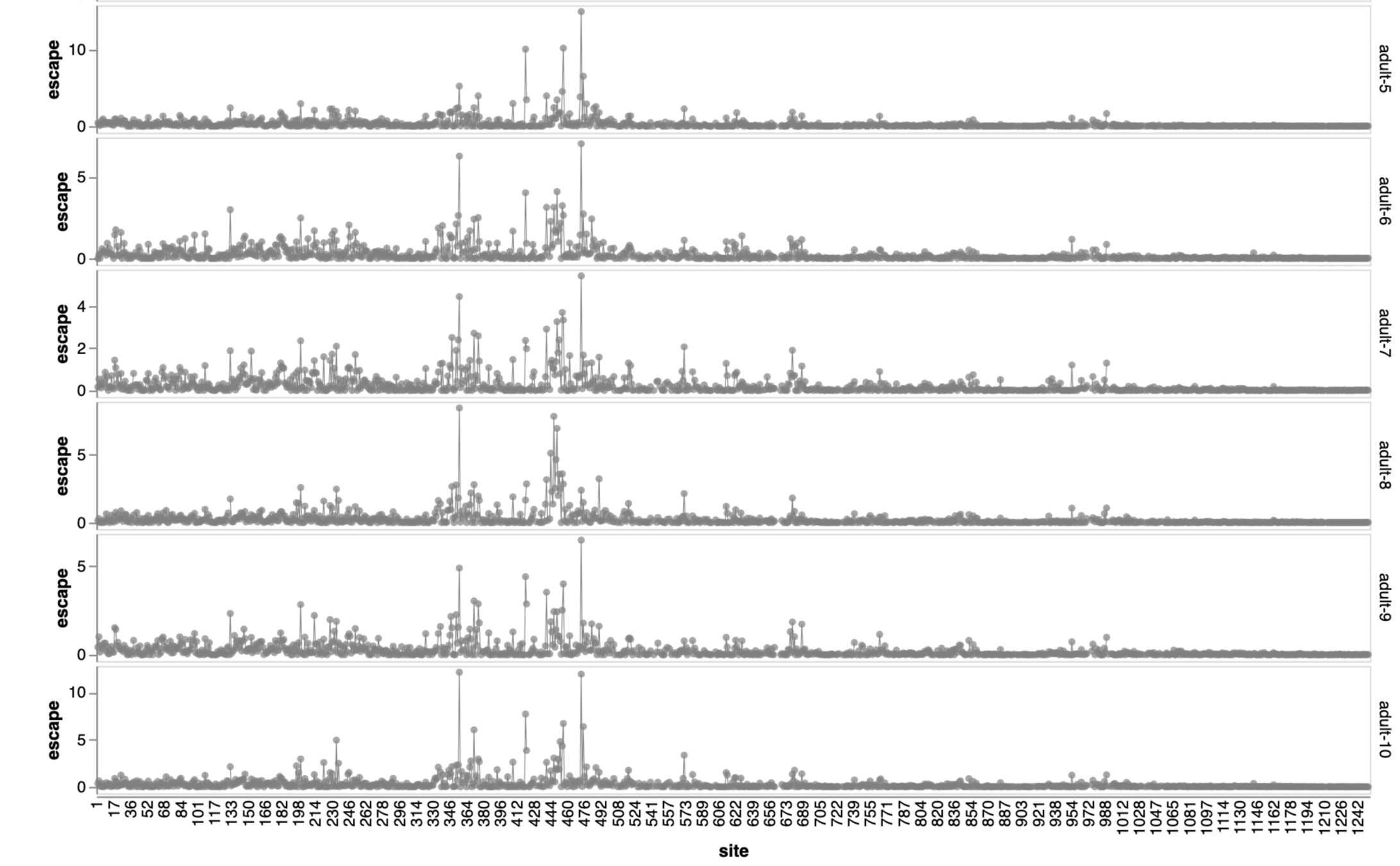
Dadonaite et al (2025); adult sera from Helen Chu's HAARVI study; infant sera from Mary Staat's IMPRINT cohort
6-12 month infants first infected by XBB*
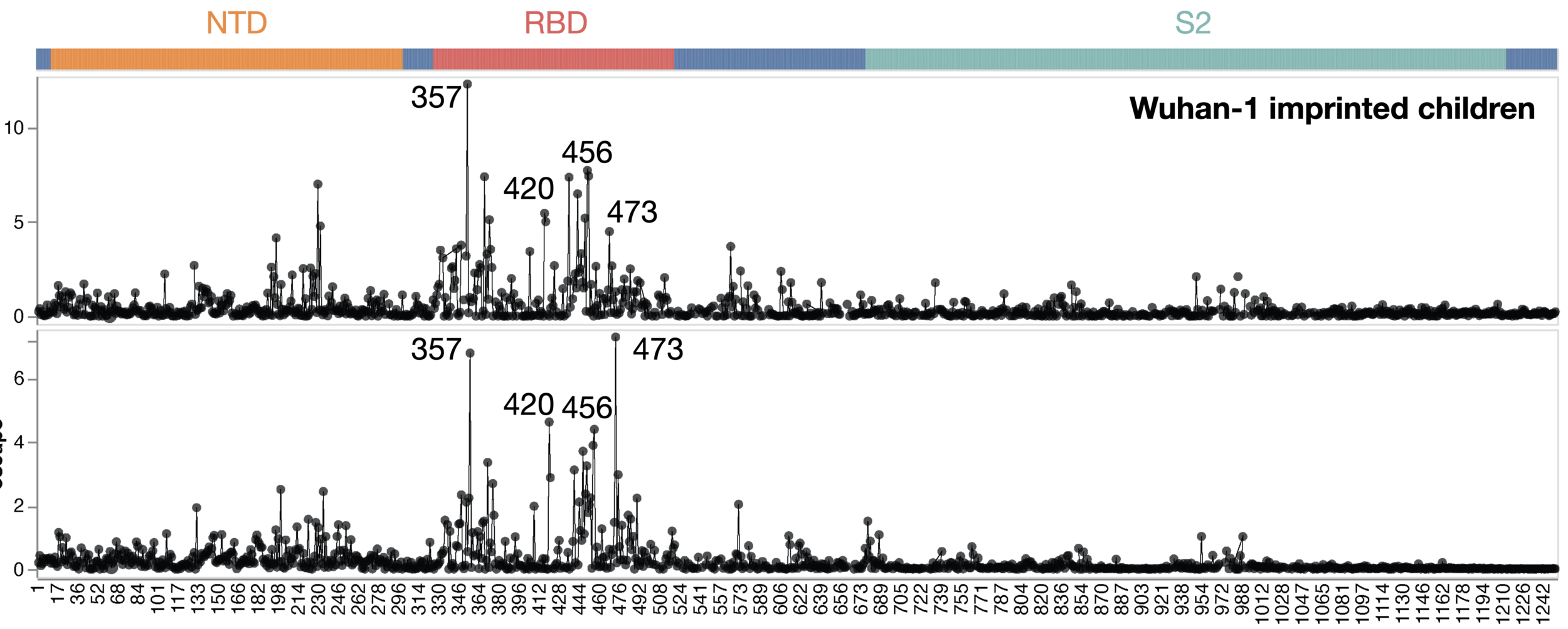
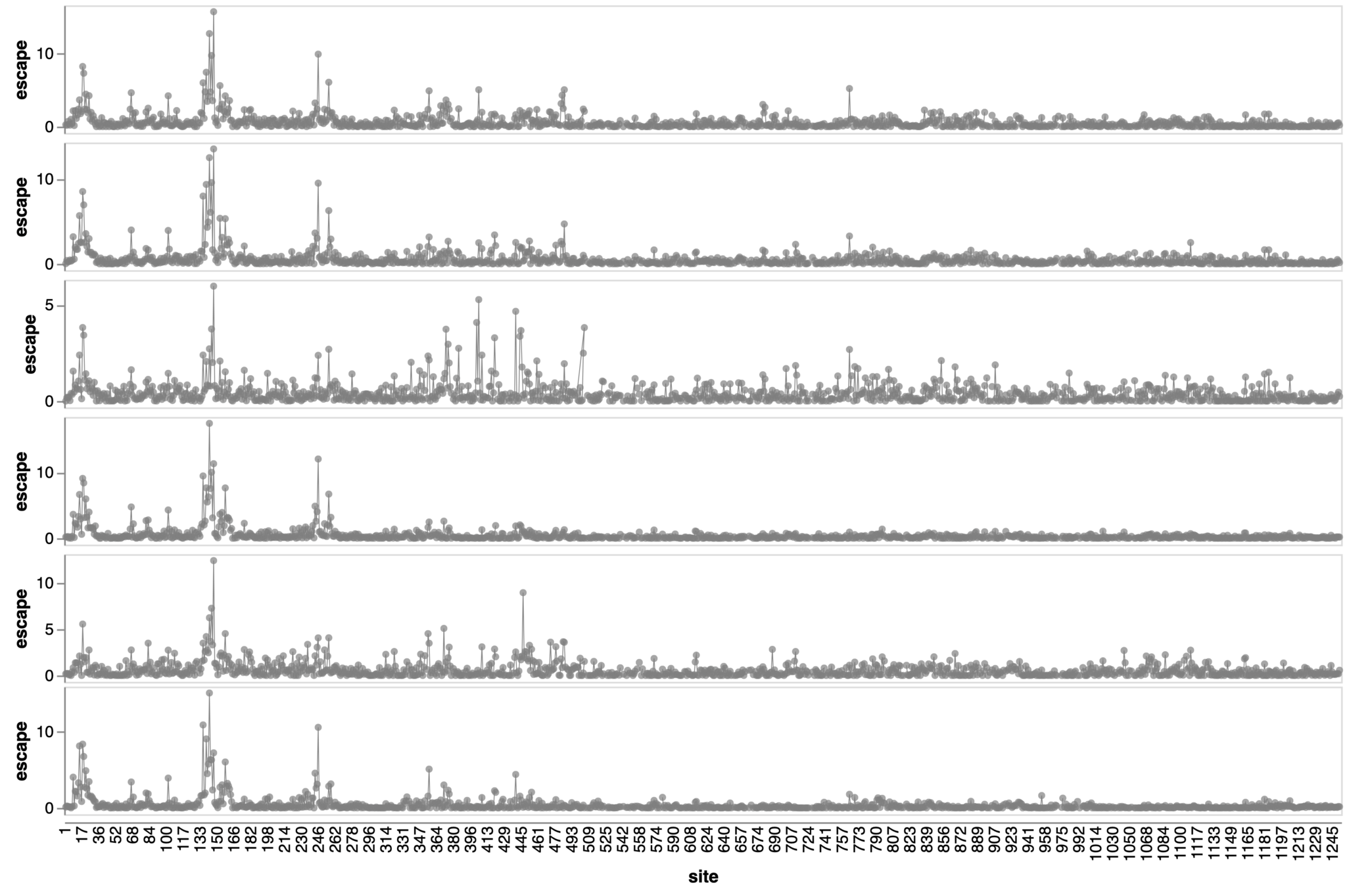
site in spike
escape caused by mutations at site
Sites of escape from sera of adults imprinted with original vaccine, then exposed to various infections and vaccinations.
adult 1
adult 2
adult 3
adult 4
adult 5
adult 6
2021
site in spike
escape caused by mutations at site
Sites of escape from infants with only single infection with XBB*
infant 1
infant 2
infant 3
infant 4
infant 5
infant 6
2023
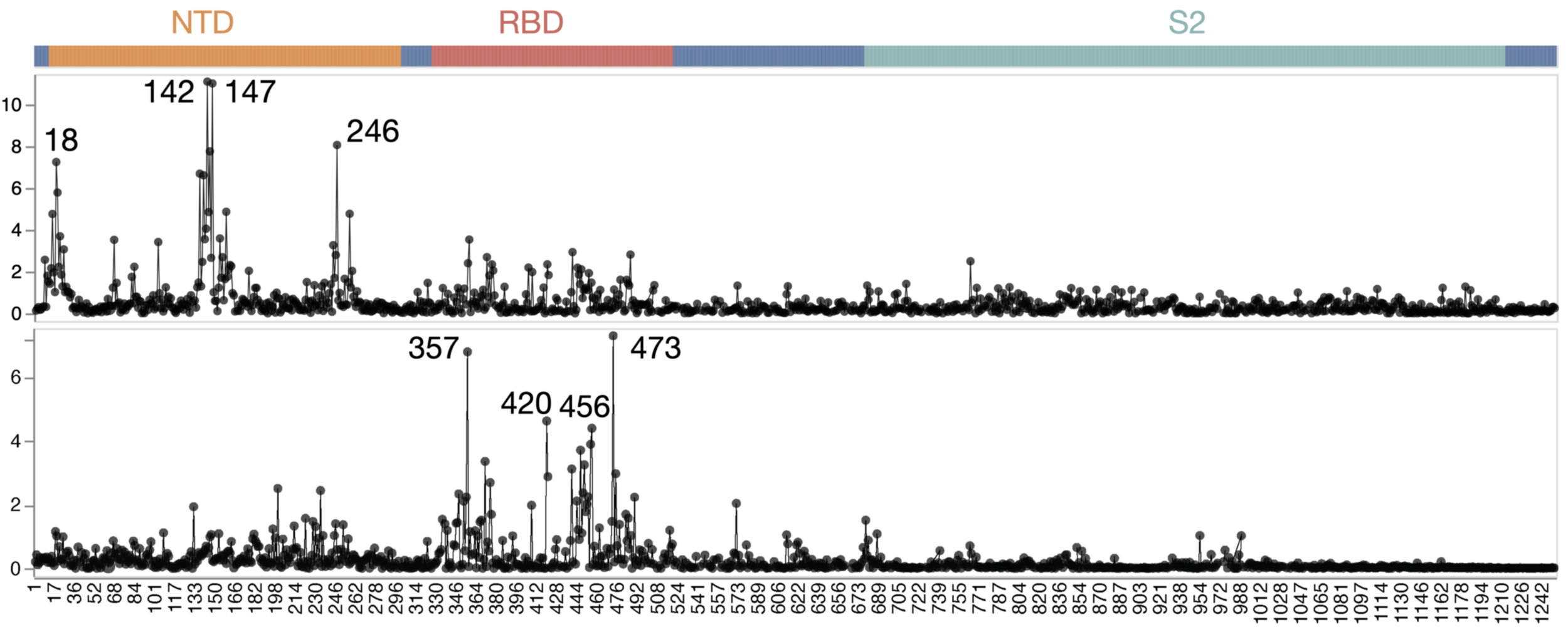
escape caused by mutations at site
site in spike
Average of sera from six infants with only a single infection by XBB* in 2023
Average of sera from ten adults imprinted by original vaccine in 2021
Human influenza evolves similarly to SARS-CoV-2, but has circulated in humans for decades, so imprinting and population immunity) are highly heterogeneous.
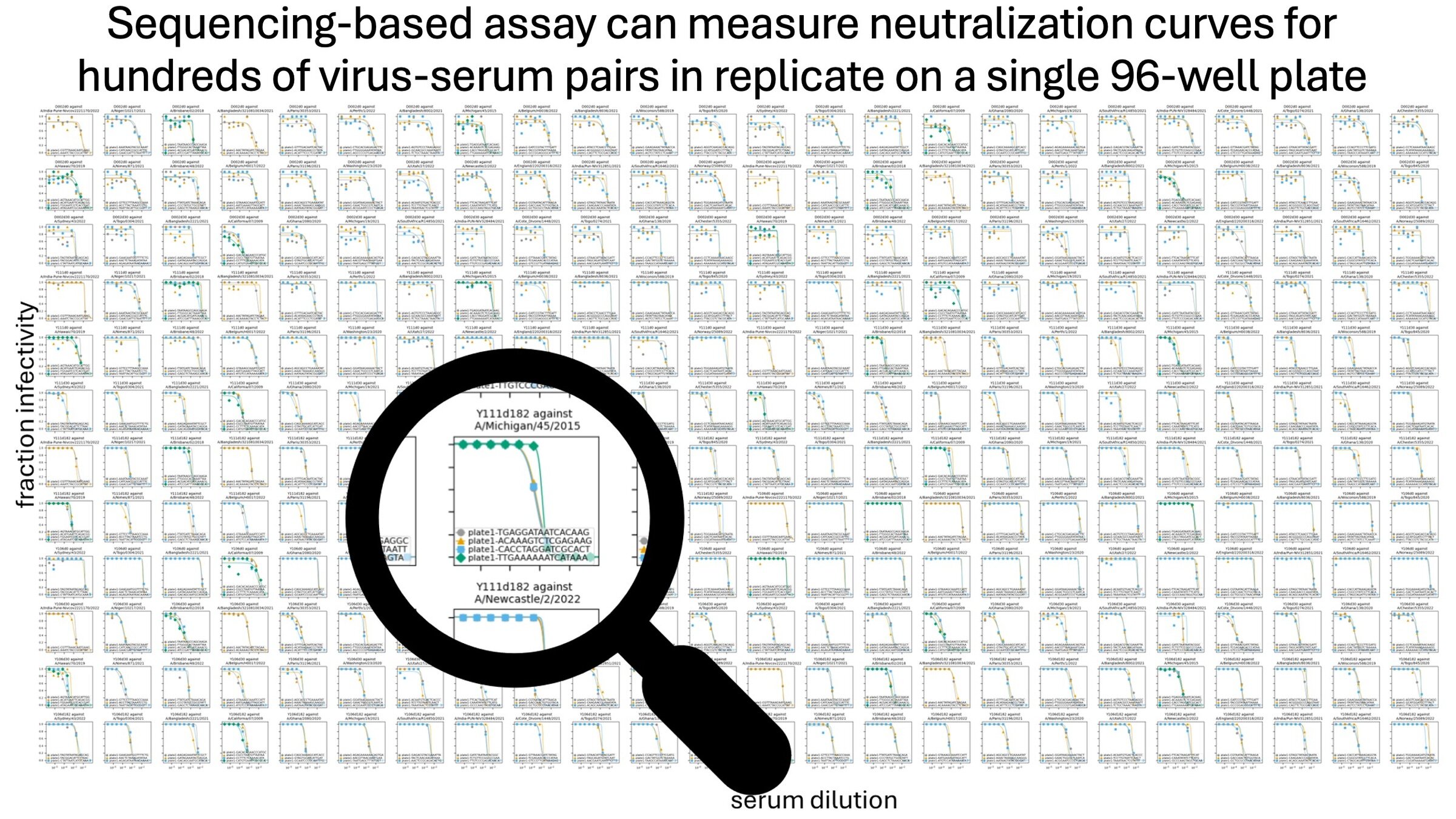
As described on the next few slides, we developed a new method to characterize immune heterogeneity for influenza by efficiently measuring neutralization of >50 viral strains by ~100 human sera.
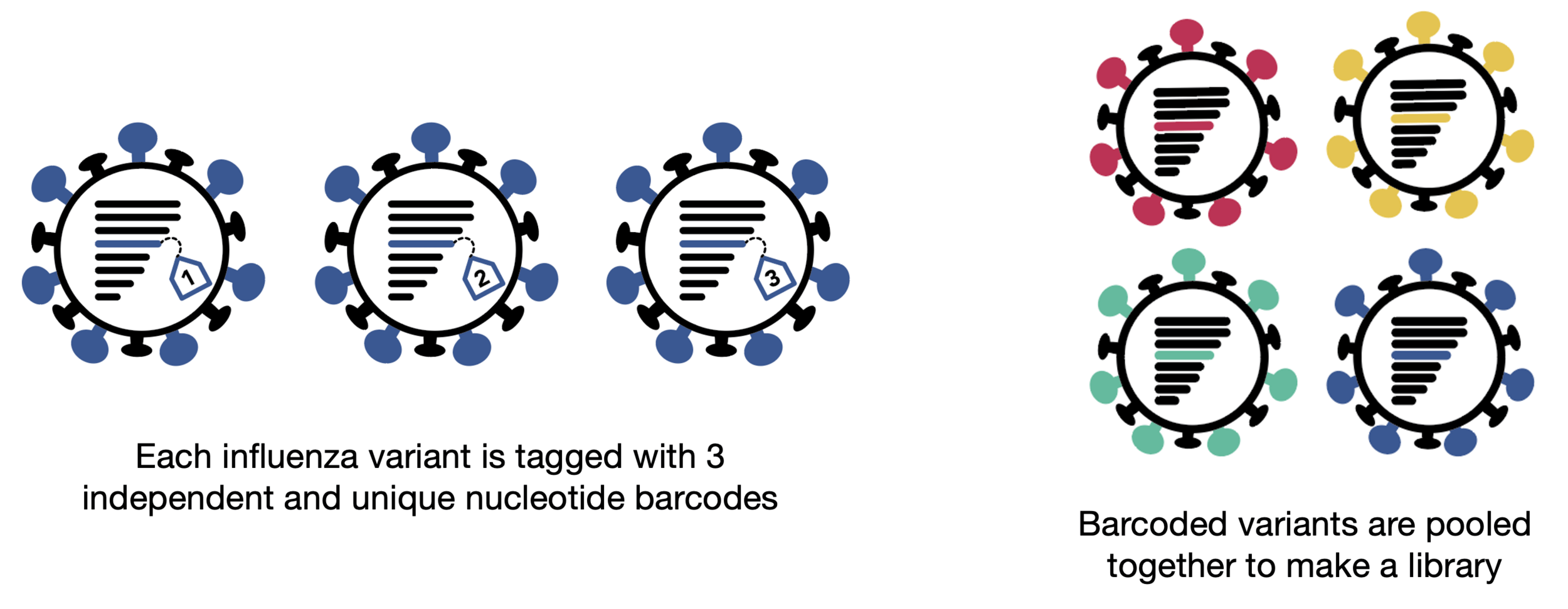
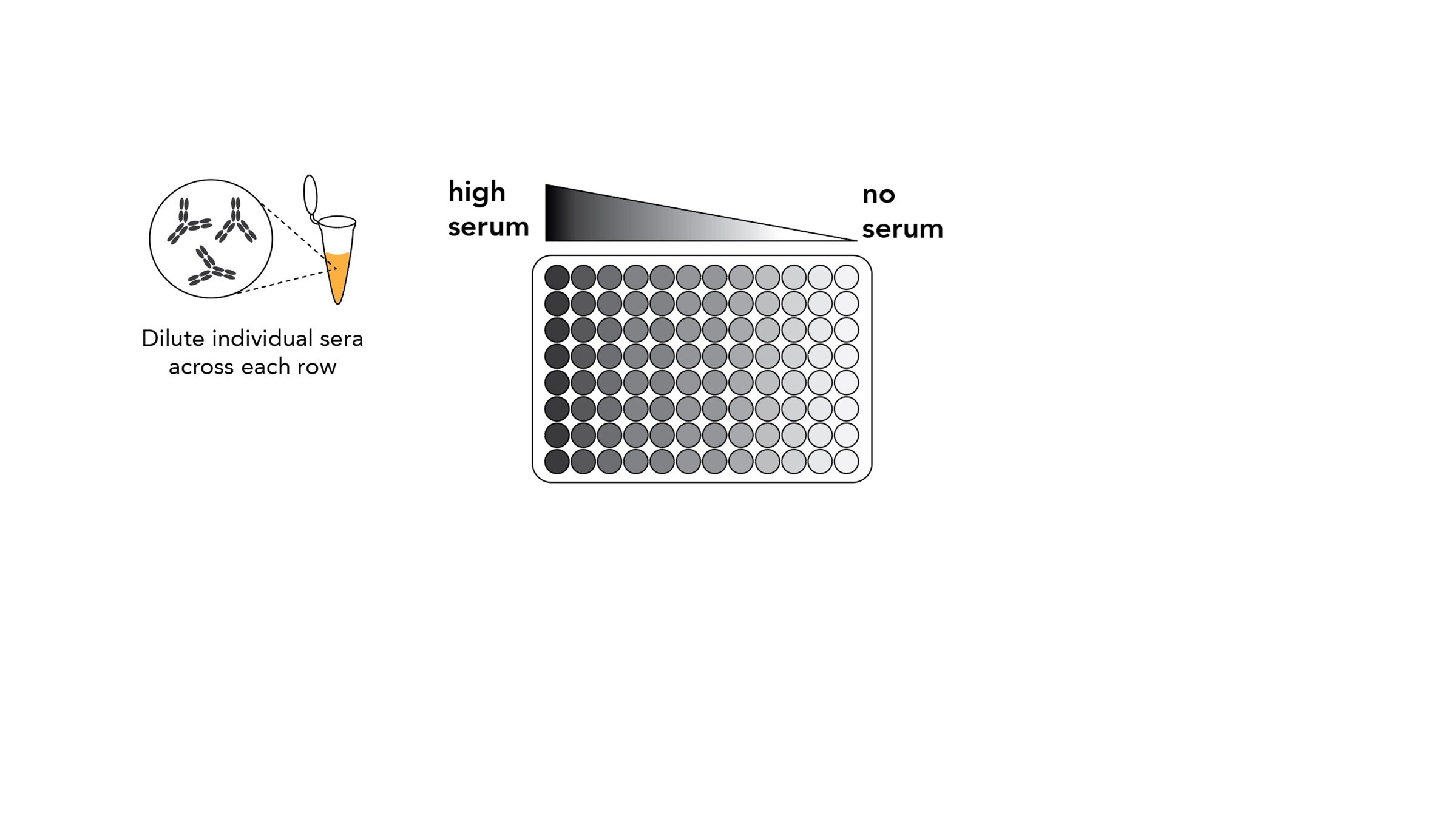
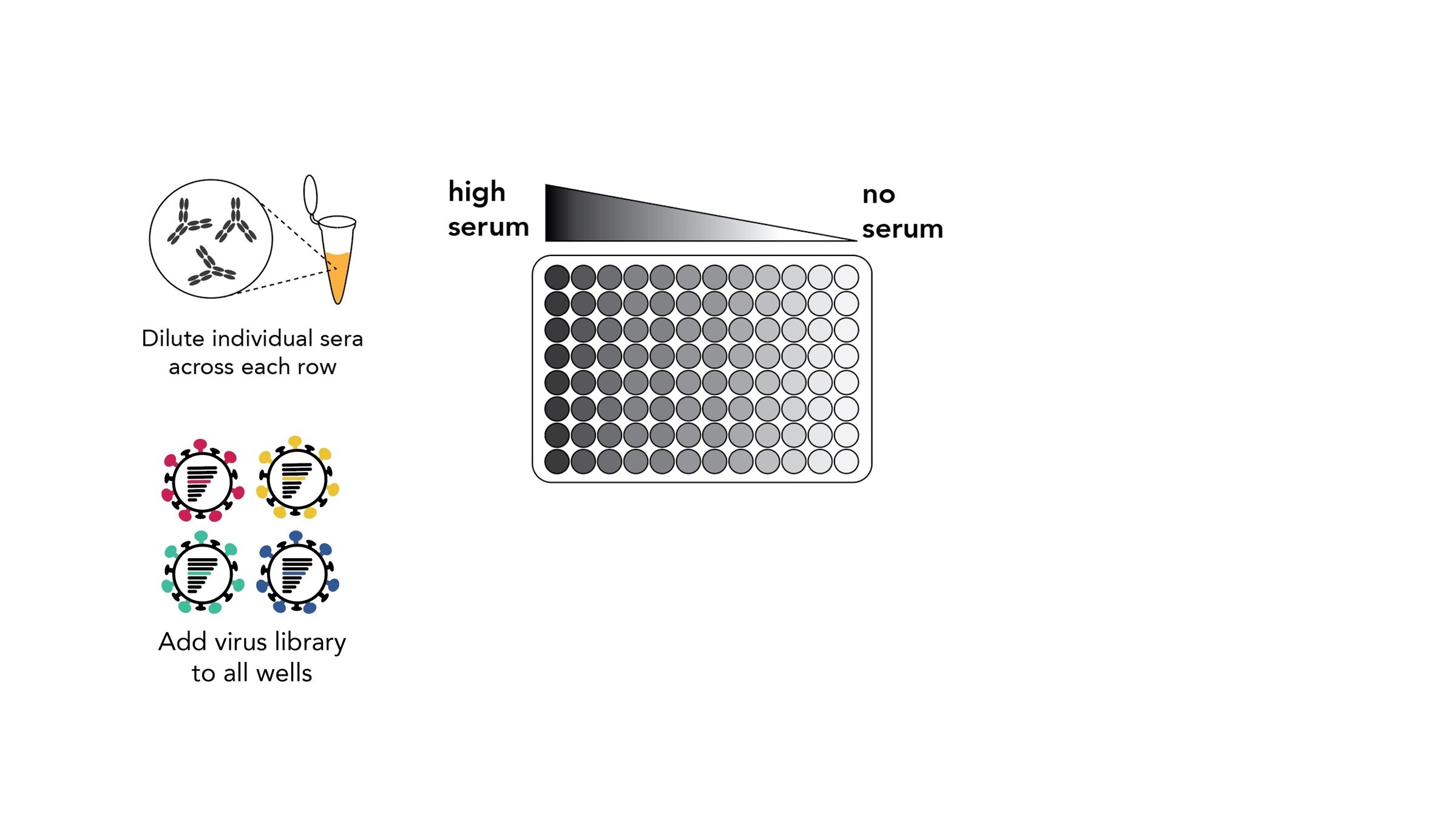
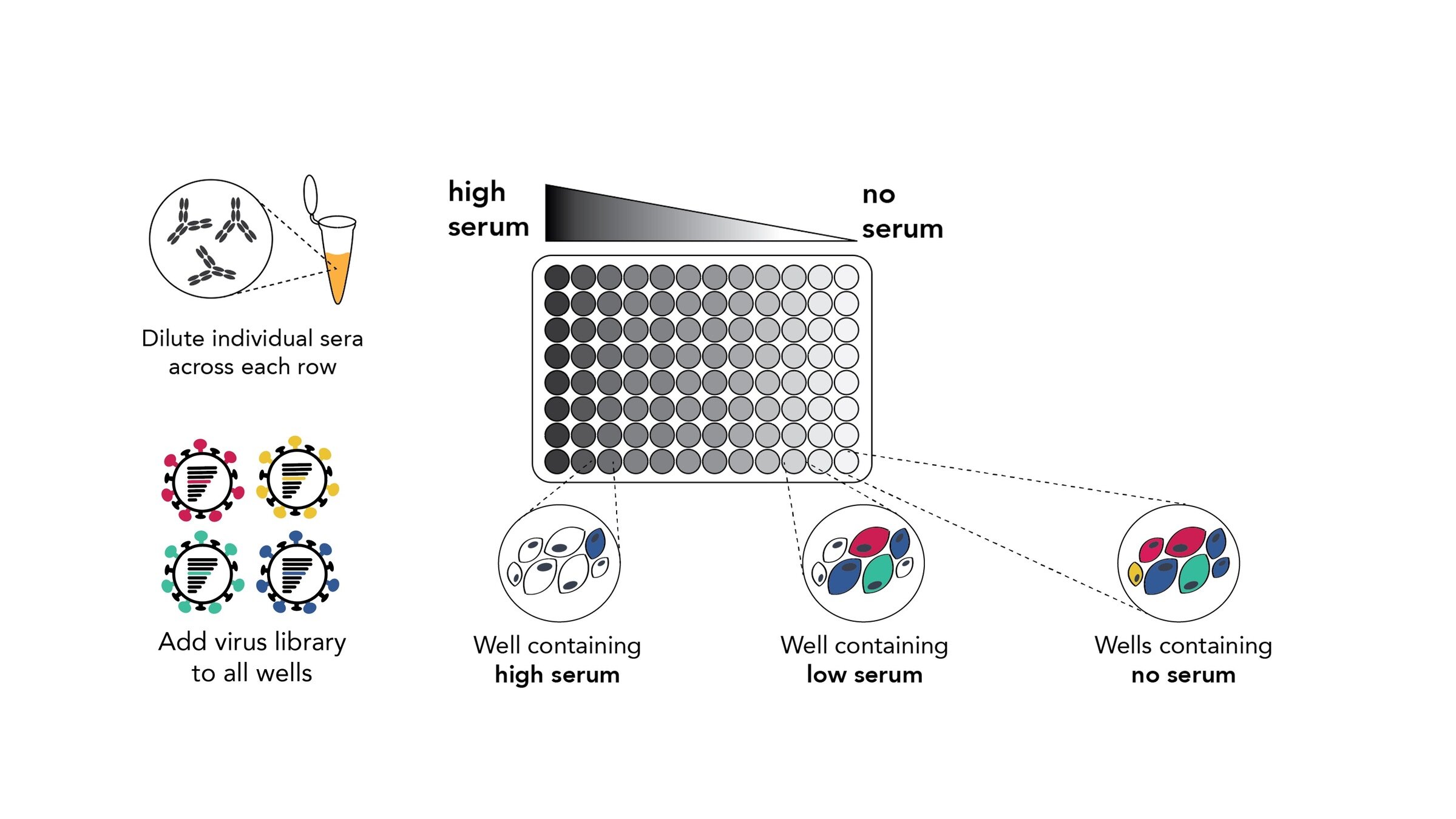
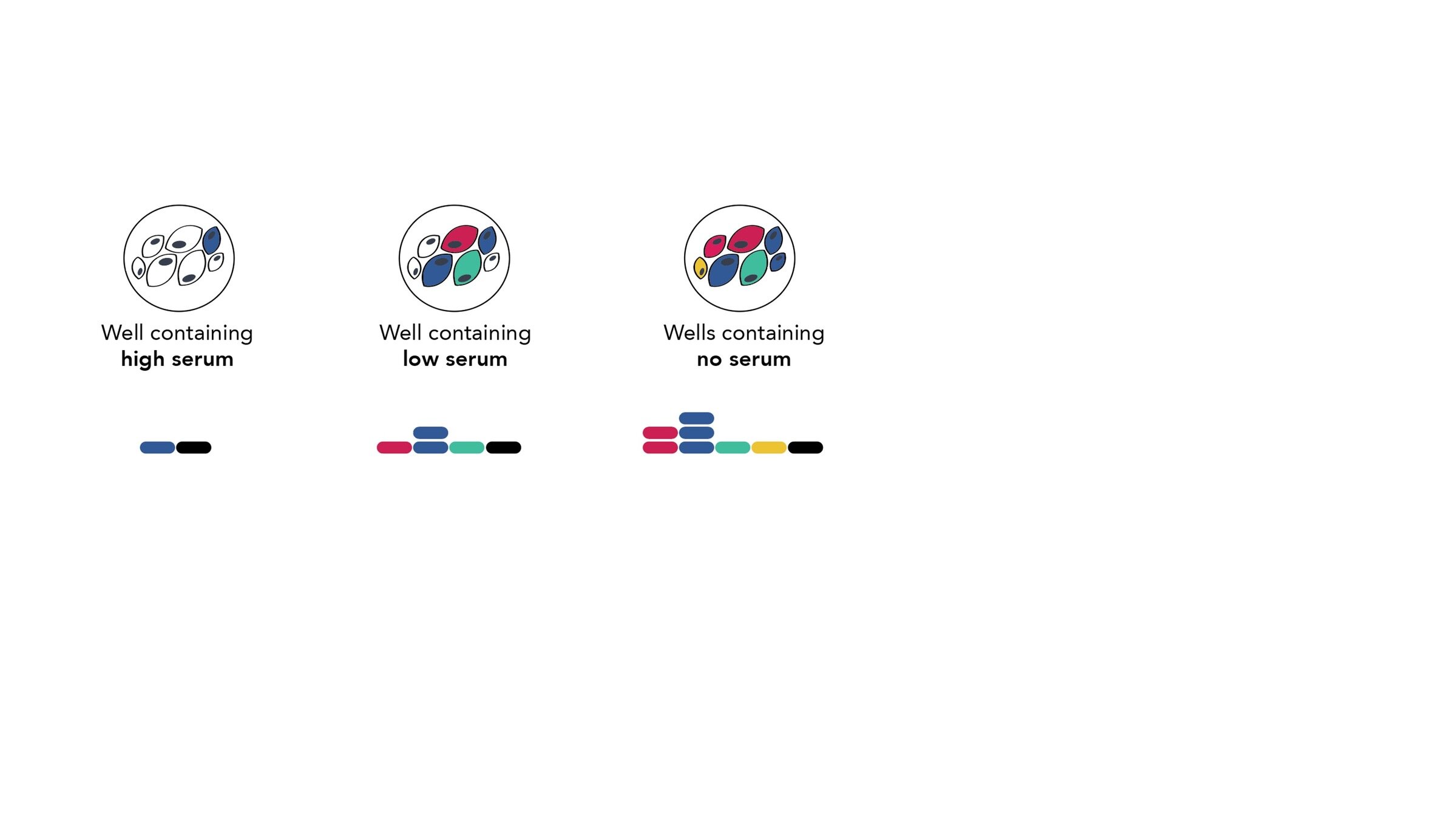
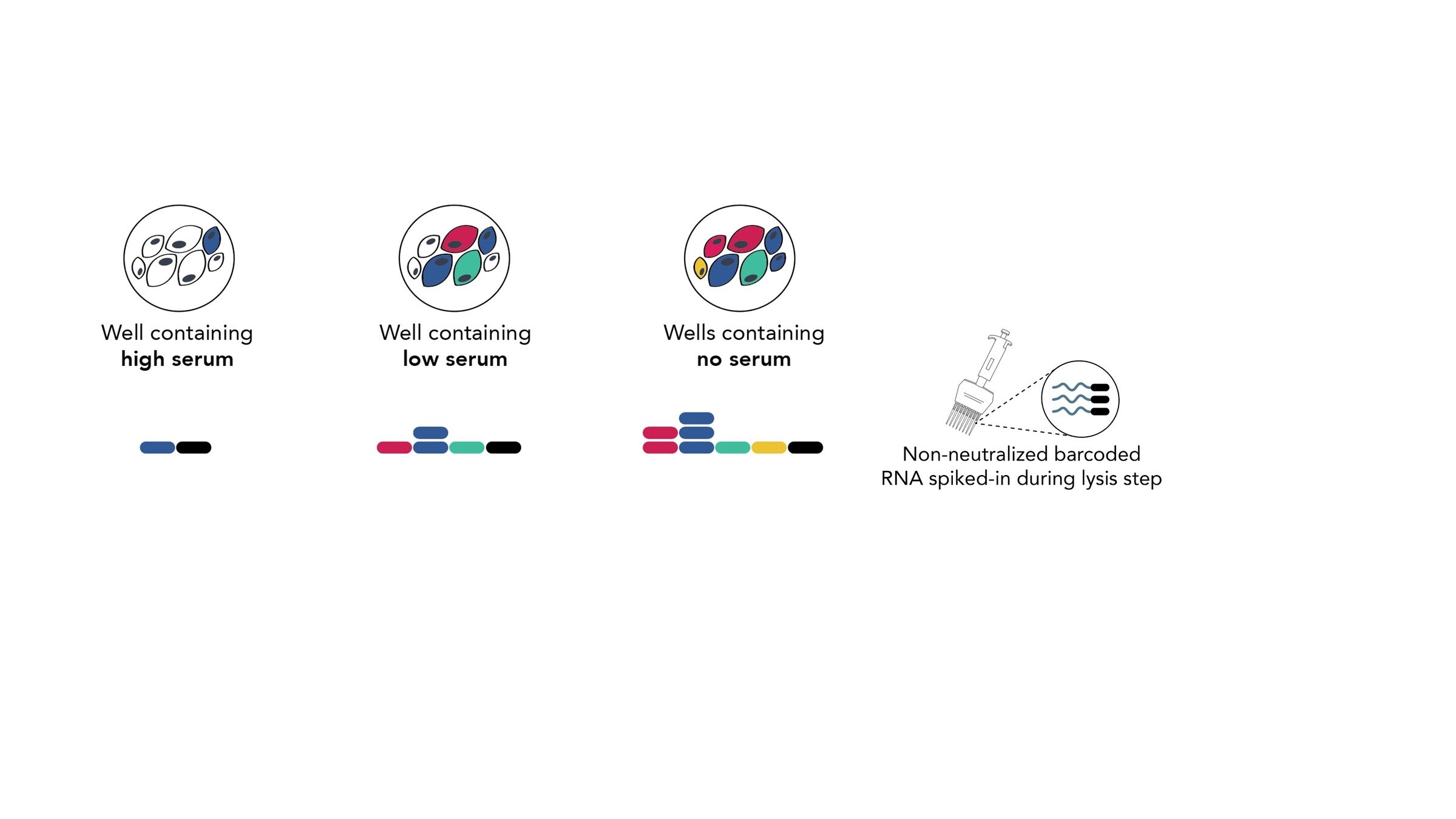
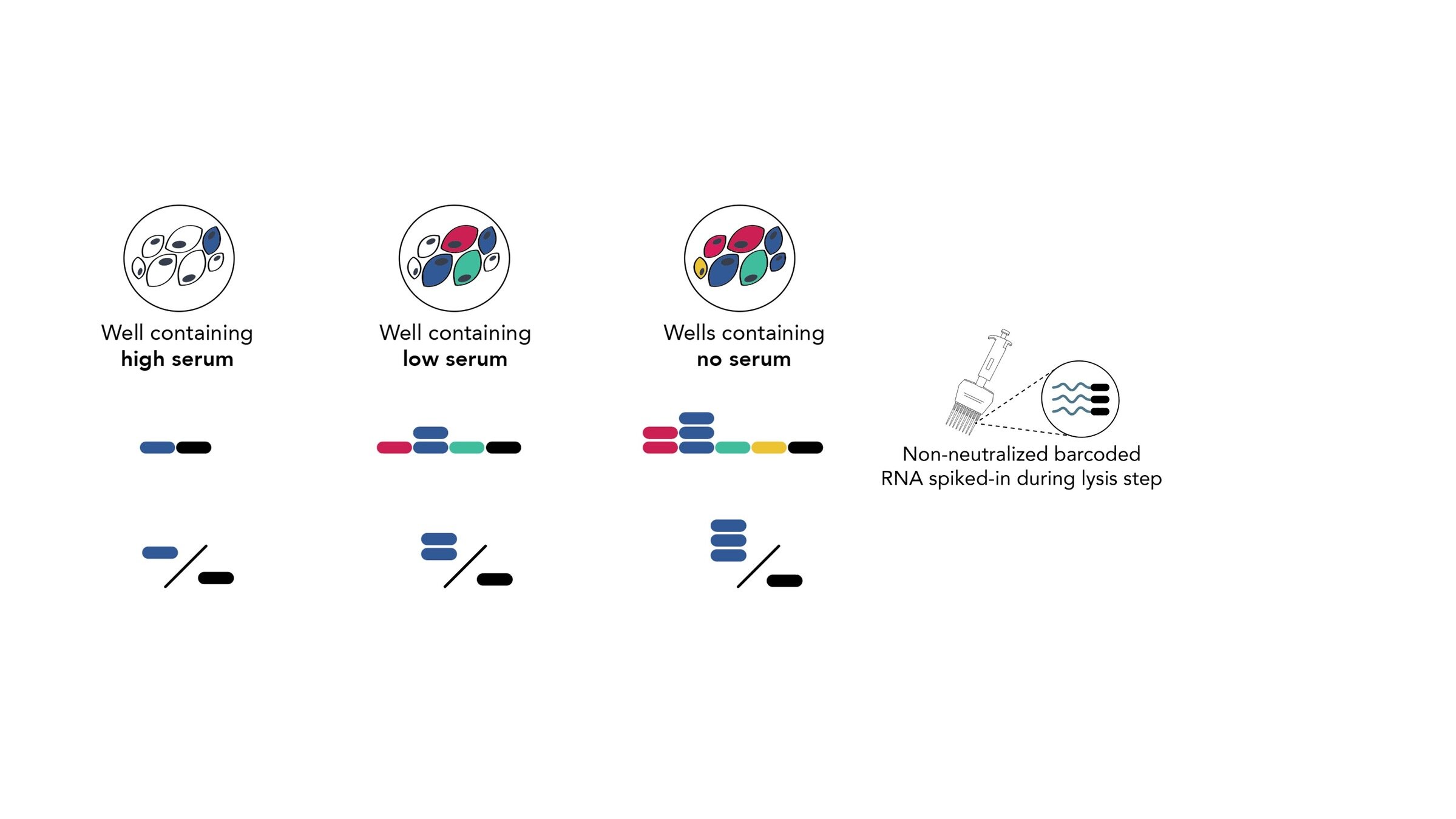
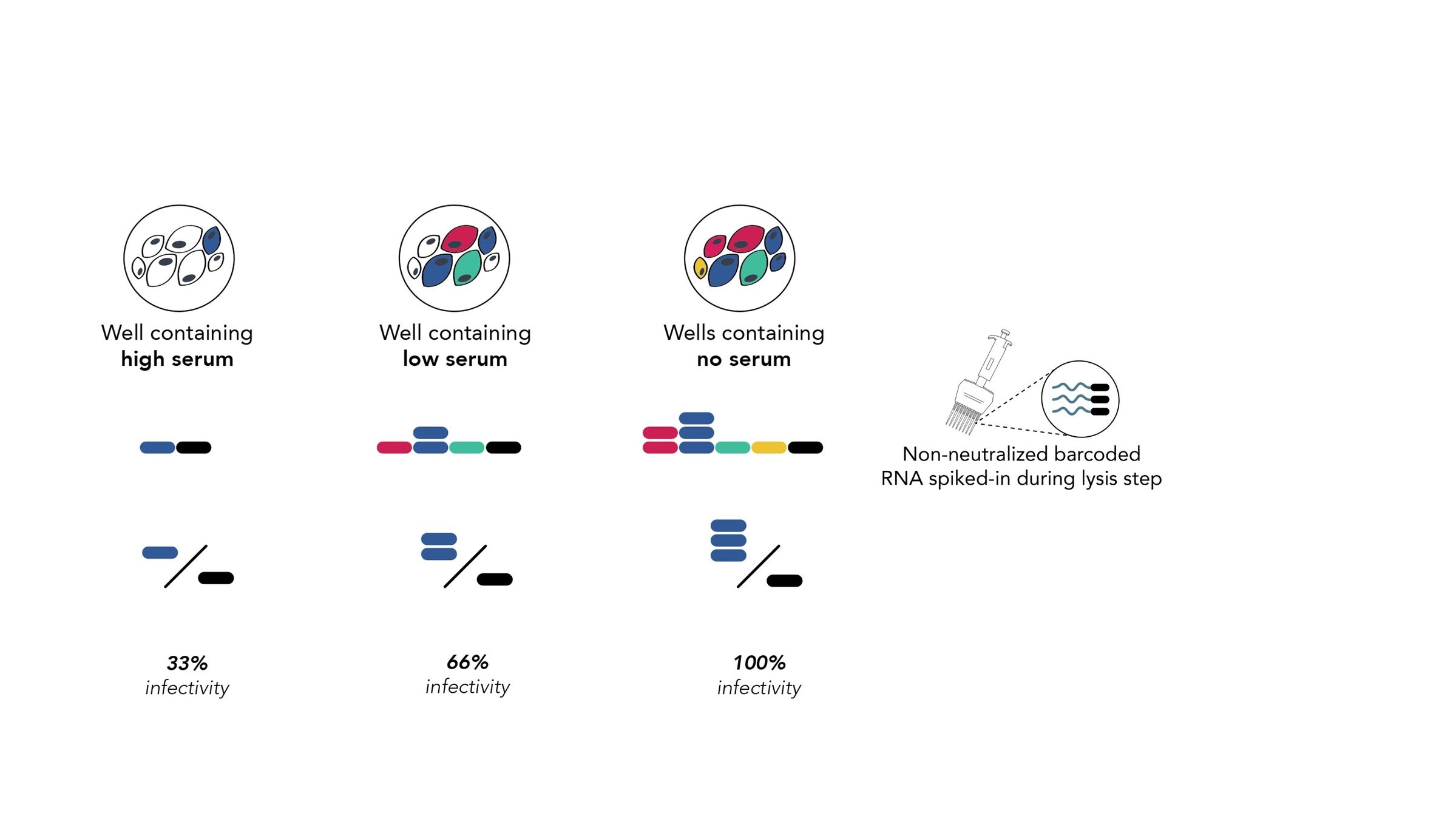
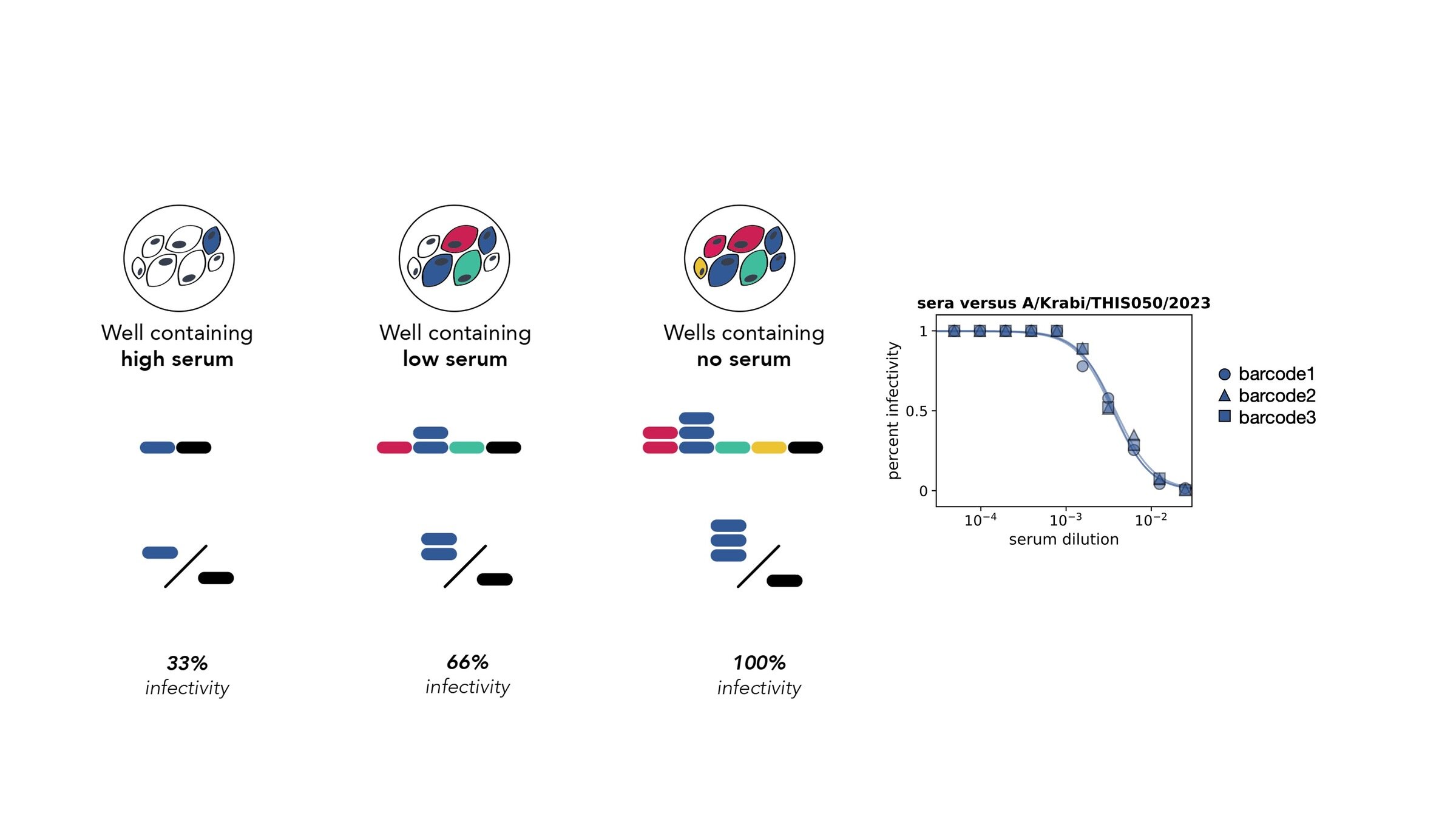
Assays measure just one virus against one serum in each row (or column) of a 96-well plate
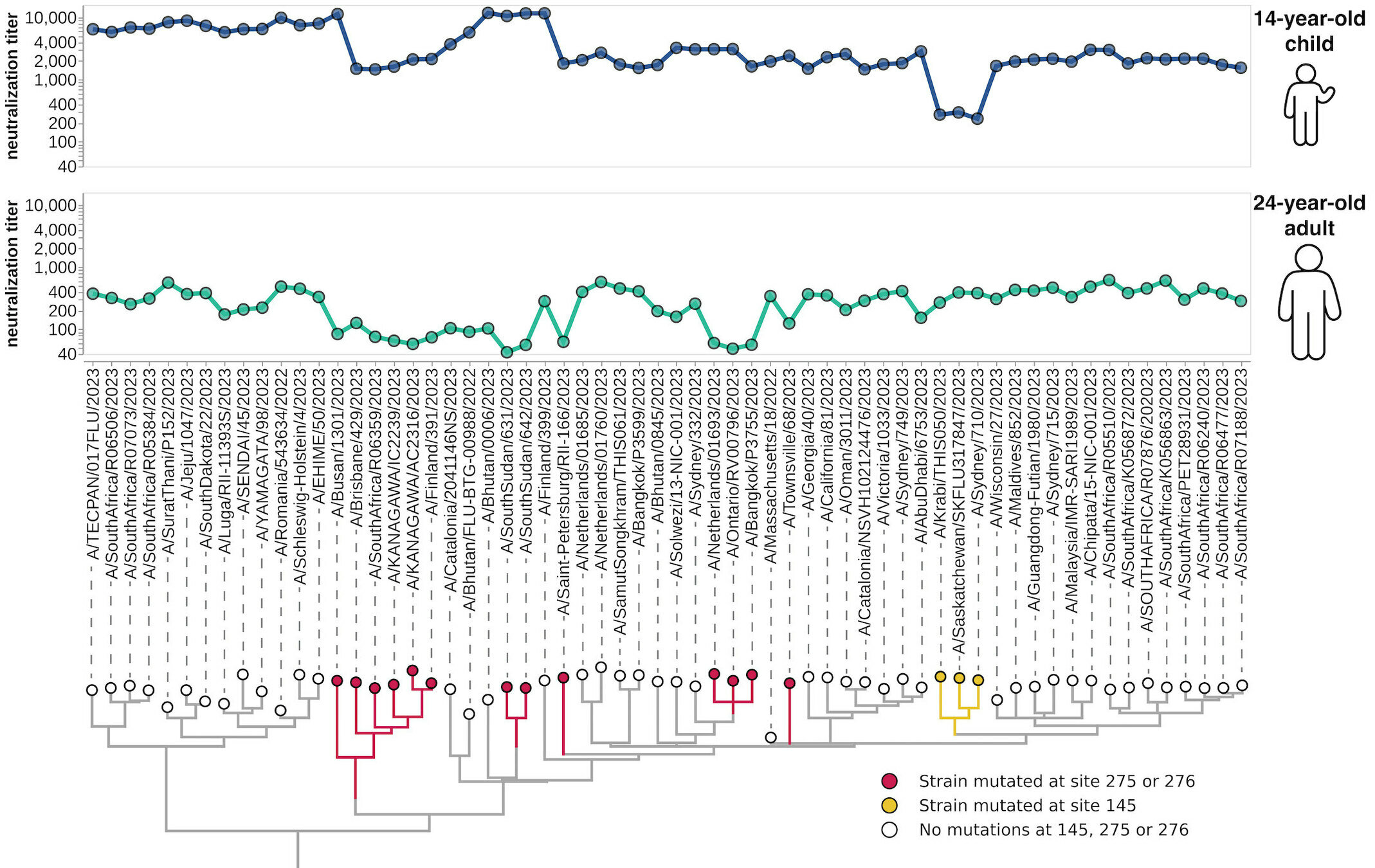
different recent H3N2 viral strains
this child has low titers to these viral strains
Unpublished data available here
sera from 95 individuals of different ages
Measured neutralization by each individual serum
Unpublished data available here
sera from 95 individuals of different ages
Measured neutralization by each individual serum
Measured neutralization by pool of all sera
(L122Q, A160T, T199I)
(L122Q, P162Q, T199I)
Bloom lab
These slides: https://slides.com/jbloom/vaxco-2025
Fred Hutch Cancer Center
Trevor Bedford
University of Washington
Helen Chu and HAARVI cohort
Neil King
David Veesler
Cincinnati Children's Hospital
Mary Staat
David Haslam
Allie Burrell
University of Pennsylvania
Scott Hensley
Seattle Children's Hospital
Janet Englund
Tyler Starr (now at Utah)
Allie Greaney (now at UCSF)
Also: Kate Crawford, William Hannon, Caelan Radford
Bernadeta Dadonaite
Rachel Eguia
Caroline Kikawa
Andrea Loes
By Jesse Bloom



