












Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
These slides at https://slides.com/jbloom/hhmi-meeting-2024
viral membrane
cell membrane
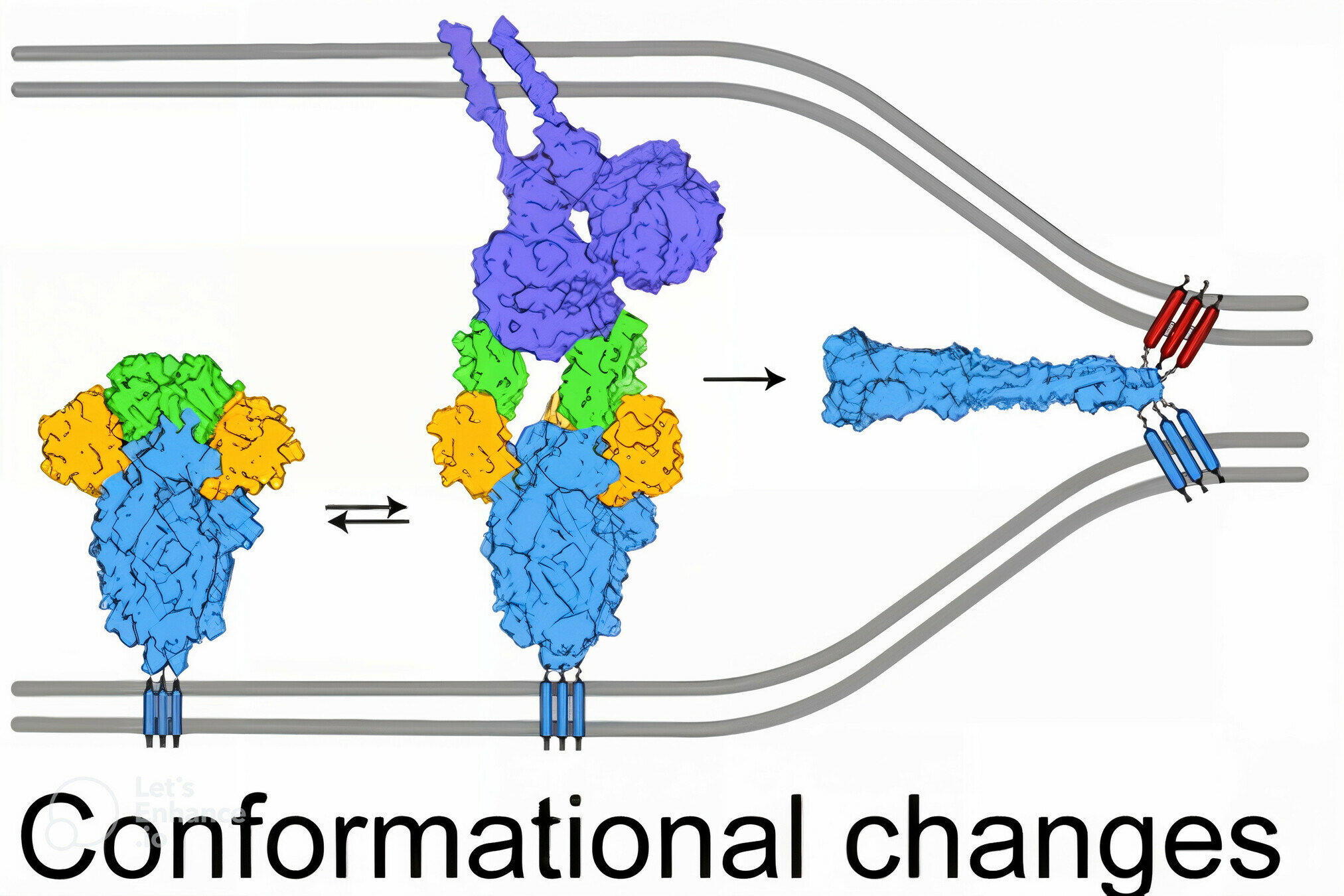
spike
spike conformational change
Image adapted from here
ACE2
antibody
Image adapted from here
RBD
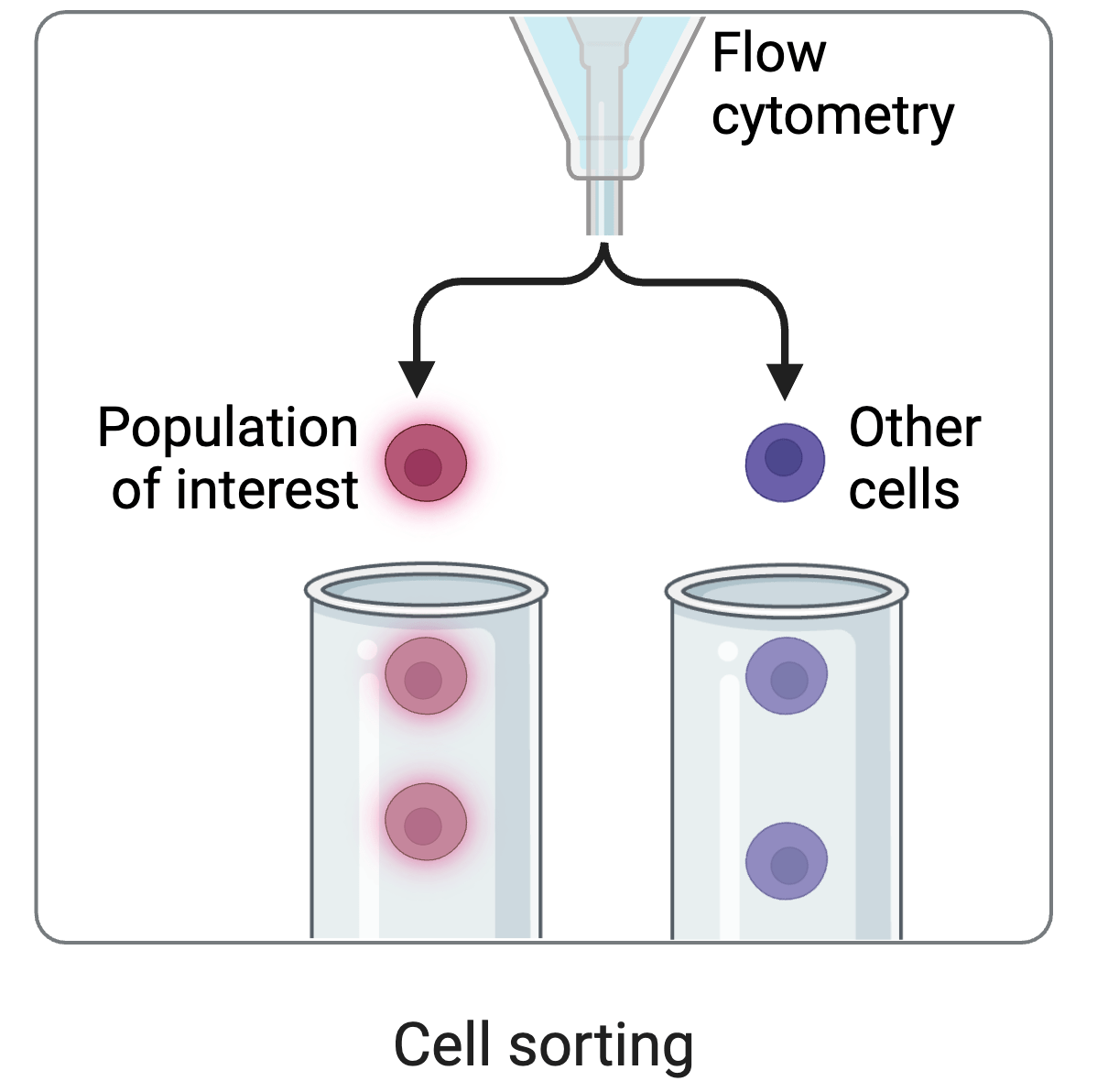
fluorescent ACE2
yeast
fluorescent tag on RBD
cell sorting
RBD
fluorescently labeled antibody
yeast
fluorescent tag on RBD
site in RBD
antibody escape
484
cell sorting
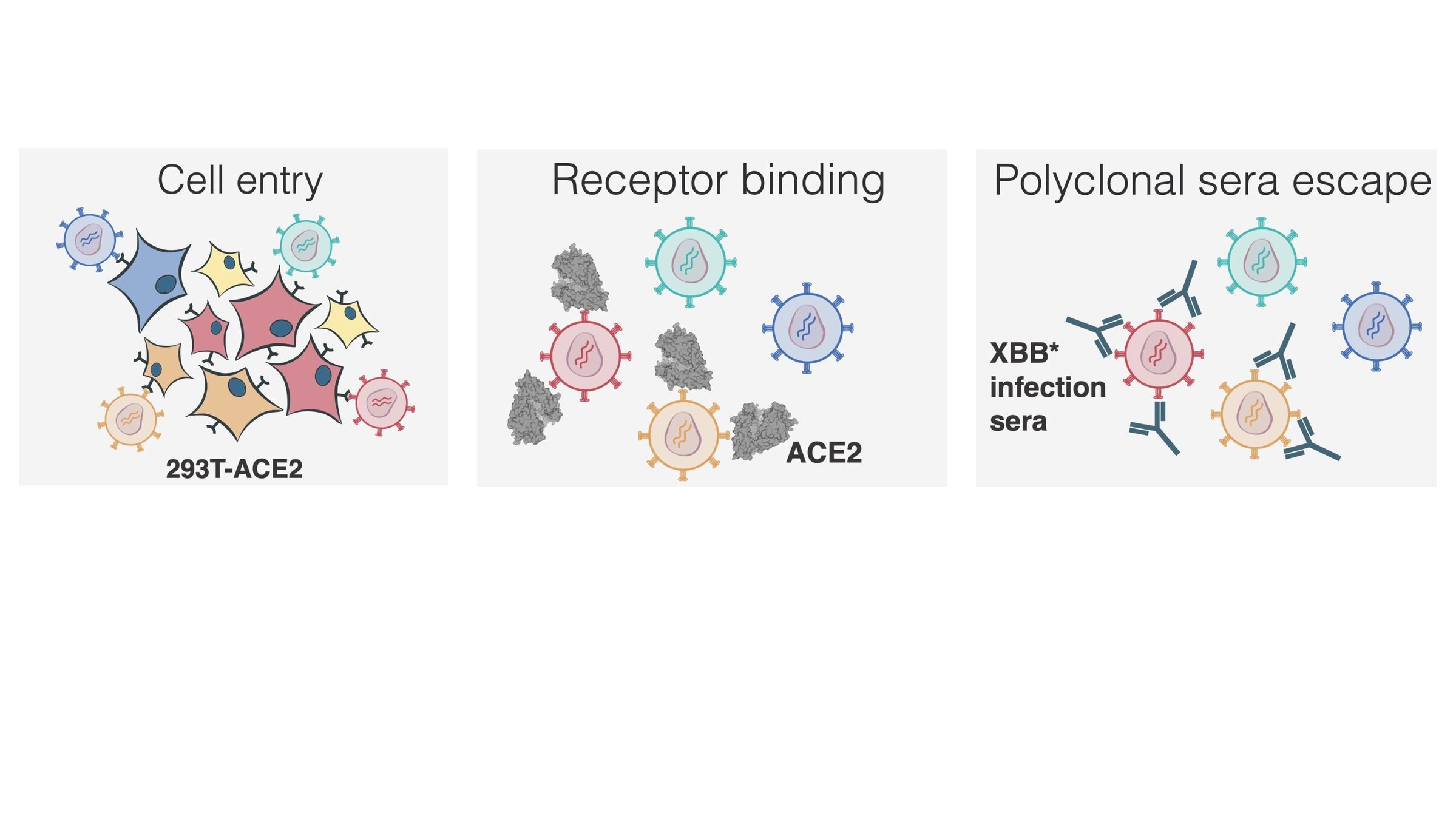
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
With Trevor Bedford & Ben Murrell
With Trevor Bedford & Ben Murrell
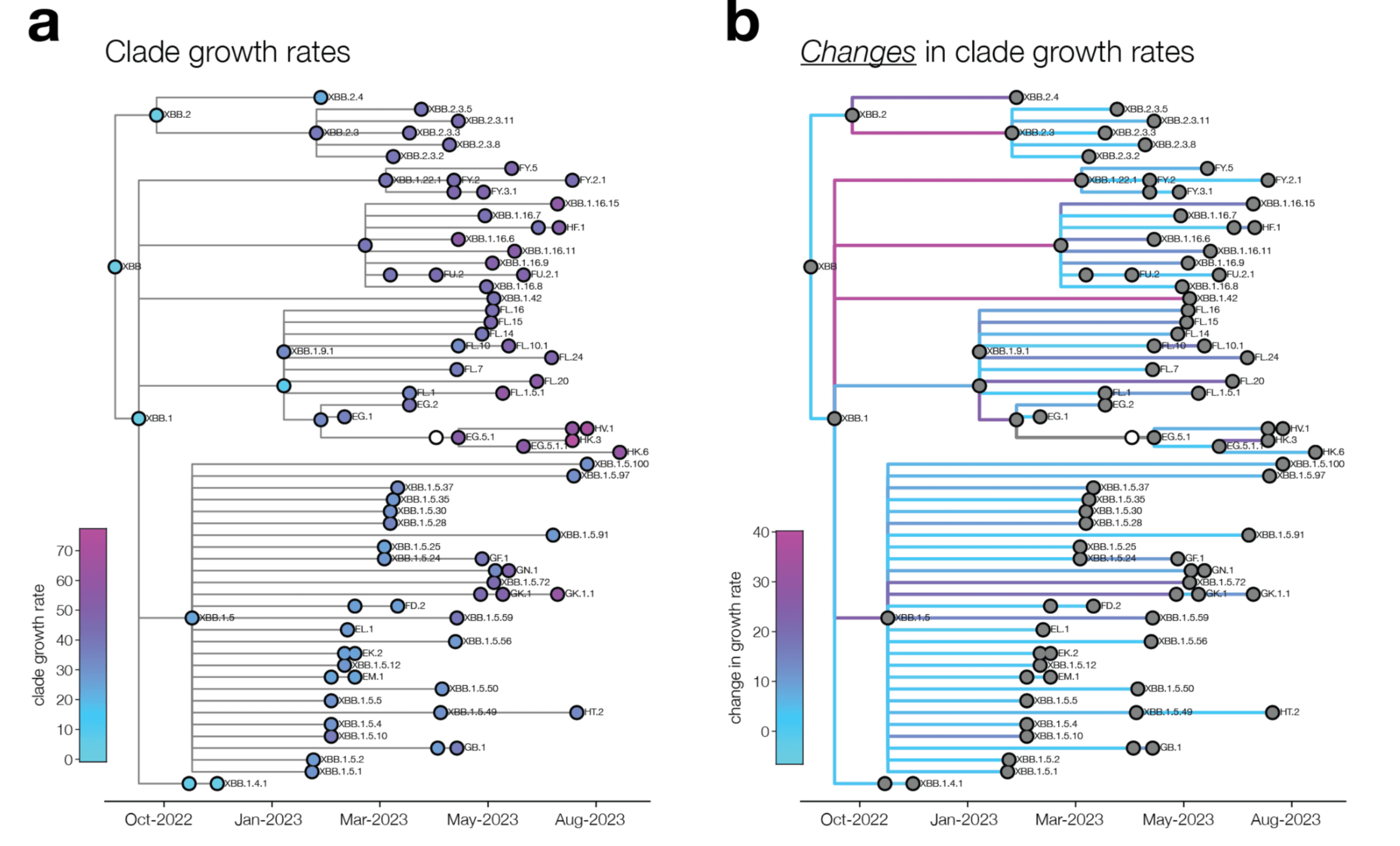
change in clade growth
clade growth
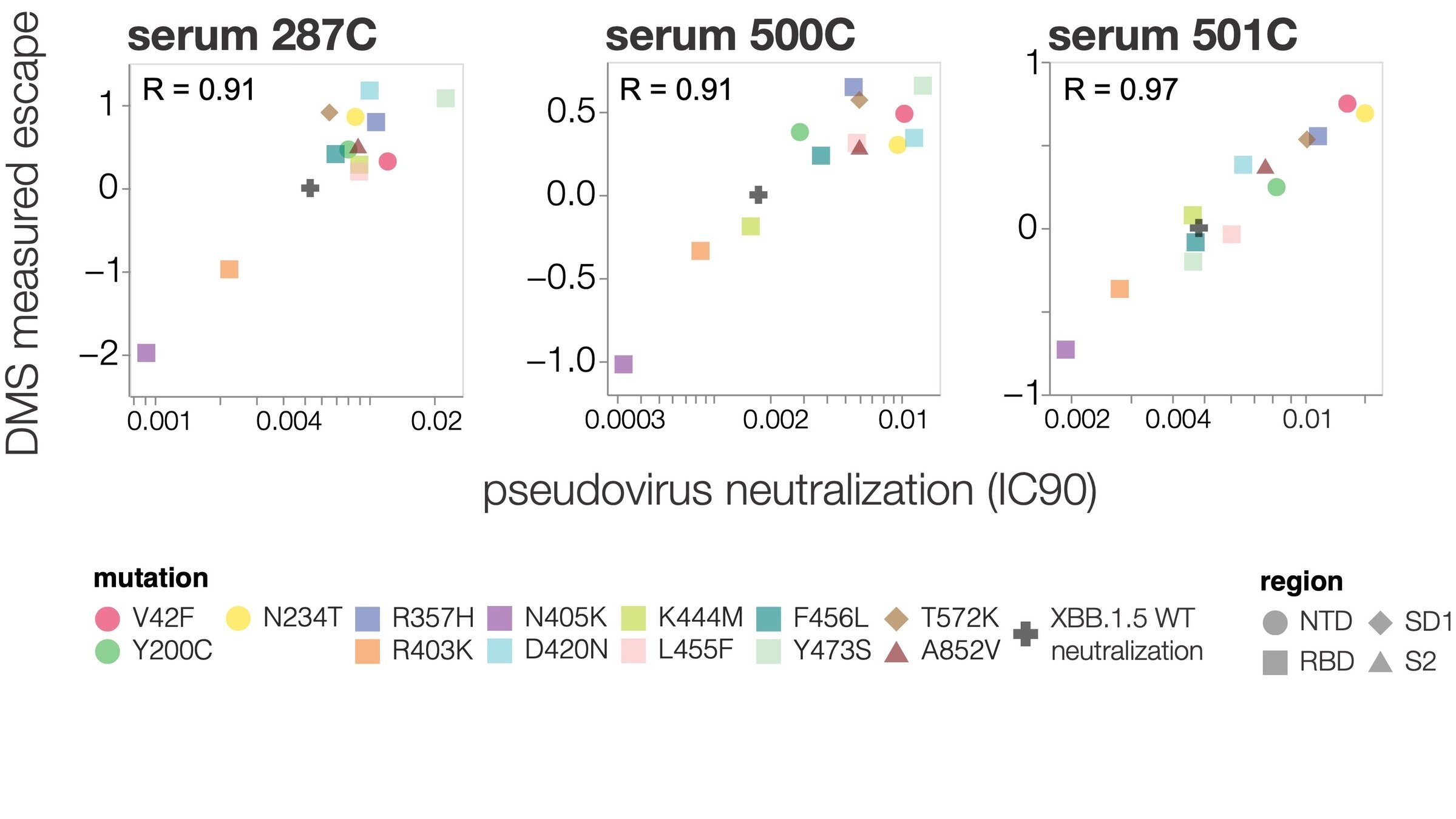
We can explain ~55% of the variance in growth of different clades, with largest fraction of variance uniquely explained by sera escape.
(L122Q, A160T, T199I)
(L122Q, P162Q, T199I)
Bloom lab
Bernadeta Dadonaite
Kate Crawford
Caelan Radford
Tyler Starr
Allie Greaney
Rachel Eguia
William Hannon
Jenny Ahn
Fred Hutch Cancer Center
Trevor Bedford
John Huddleston
University of Washington
Helen Chu and HAARVI cohort
Neil King
David Veesler
Pirbright Institute
Thomas Peacock
University of Pennsylvania
Scott Hensley
Louise Moncla
Jordan Ort
St Jude Children's Hospital
Richard Webby
By Jesse Bloom
From molecular phenotype to virus evolution: deep mutational scanning of the SARS-CoV-2 spike



